Genes & Cancer

Givinostat, a type II histone deacetylase inhibitor, induces potent caspase-dependent apoptosis in human lymphoblastic leukemia
Ying Li1,*, Kevin Zhao2,*, Chenjiao Yao1,*, Samir Kahwash3, Yan Tang2, Guojiuan Zhang2, Kara Patterson2, Qi-En Wang4 and Weiqiang Zhao2
1 The Third Xiangya Hospital of Central South University, Hunan, China
2 Department of Pathology, The Ohio State University Wexner Medical Center, Columbus, OH, USA
3 Department of Pathology, Nationwide Children’s Hospital, Columbus, OH, USA
4 Department of Radiology, The Ohio State University Wexner Medical Center, Columbus, OH, USA
* These authors contribute equally to the manuscript
Correspondence to: Weiqiang Zhao, email: [email protected]
Keywords: acute lymphoblastic leukemia, givinostat, apoptosis, p53, BCR-ABL
Received: May 24, 2016
Accepted: September 25, 2016
Published: September 28, 2016
ABSTRACT
Unlike chronic myeloid leukemia, patients with acute lymphoblastic leukemia (ALL) with Philadelphia chromosome (Ph+) do not respond well to Imatinib or tyrosine kinase inhibitors (TKI). In addition, TKI might induce resistant mutations in kinase domain (KD) of ABL in patients with relapsed diseases. Of the histone deacetylase (HDAC) inhibitors, suberoylanilide hydroxamic acid (SAHA) has shown to induce potent cytotoxicity on acute myeloid leukemia cell lines but Givinostat effect on acute lymphoblastic leukemia (ALL) has not been reported. We investigated if Givinostat could exert similar inhibitory effect on SUP-B15, an established B-cell ALL with Philadelphia chromosome (Ph+). Two Ph+ leukemia cell lines, SUP-B15 and an AML cell line K562 were studied in parallel for their responses to Givinostat. Mutation status of TP53 genes was also examined to correlate cellular proliferation and apoptosis. Givinostat significantly inhibited cell proliferation of SUP-B15 (IC50:0.18±0.03µM) and simultaneously inhibited BCR-ABL signal pathway. A remarkable apoptosis was induced by 0.25µM Givinostat in SUP-B15 along with the activation of caspase cascades and increased expression of p21. These inhibitory and proapoptotic effects were not observed in K562 simultaneously treated with Givinostat. Finally our studies showed that TP53 mutation status might determine responder or non-responder to Givinostat in these two Ph+ leukemia cell lines.
INTRODUCTION
The BCR-ABL1 fusion genes resulted from the translocation of t(9;22)(q34;q11) or Philadelphia Chromosome (Ph+) are found in virtually all chronic myelogenous leukemia (CML), one third of adult lymphoblastic leukemia (ALL), and occasionally in acute myeloid leukemia (AML) [1-2]. The chimeric BCR-ABL proteins constitutively possess tyrosine kinase activities which are postulated to be responsible for the development of leukemia via activating the Ras and mitogen-activated protein kinase pathway (RAS-MAPK), Janus-kinase (JAK)-signal transducer and activator of transcription pathways (JAK-STAT), and bcl-2/Bad/Bcl-xL anti-apoptosis signal pathway to promote cell proliferation, antiapoptosis, and genomic instability [1-3].Tyrosine kinase inhibitors (TKI) for BCR-ABL, such as Imatinib and Dasatinib, have achieved great success in treatment of CML [4-5].One of the mechanisms of TKIs induced apoptosis in K562 AML cells is proposed by trapping BCR-ABL in the nuclei of leukemic cells [6].In contrast, patients with Ph+ ALL do not respond well to these target medicines [7]. In addition, a previous study demonstrated that TKI might induce resistant tyrosine kinase domain (KD) mutations in ABL in the vast majority of patients with recurrent disease that received TKI therapy [8].Therefore, efforts on finding novel therapeutic agents and approaches will benefit these patients.
Acetylation and deacetylation of N-terminal tails of histones regulated by histone acetyltransferases or histone deacetylases (HDACs) result in remolding of chromatin which selectively turn on or off the genes of the interest, hence being ideal epigenetic targets by medicine, namely HDAC inhibitors [9-11].Suberoylanilide Hydroxamic Acid (SAHA) also known as Vorinostat is a prototype of HDAC inhibitor in the treatment of both solid and hematologic malignancies [12-13].Givinostat (ITF2357), similar to SAHA with combined Class I+II HDAC inhibitory effects has shown anti-inflammatory properties at low nanomolar concentrations in humans, and proven to be a safe oral medication [14].Previous study showed its anti-neoplastic activities against cells with JAK2 V617F mutation, a hallmark for human myeloproliferative neoplasm (MPN) [15-17], and on a T-cell ALL [18]. In this study we demonstrated that Givinostat also had potent anti-leukemic effects in SUP-B15, a Ph+ B-cell ALL cell line resistant to TKIs.
RESULTS
Givinostat induced anti-proliferation of ALL cells and inhibited BCR-ABL signal pathway
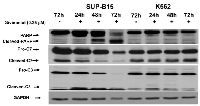
We performed dose-effect study of Givinostat at 48hrs on K562 and SUP-B15. Givinostat significantly suppressed the proliferation of SUP-B15 starting at 0.10 µM and reached to a plateau at 0.25 to 0.50µM. Givinostat had minimum inhibition on K562 except at highest concentration tested, 0.5µM (Figure 1A). The IC50 of Givinostat treatment at 48 hours was determined from cell survival plots (SigmaPlot). The IC50 of Givinostat on SUP-B15 was 0.18±0.03µM while on K562 was 4.6±0.35µM, and the difference was statistically significant (P < 0.0039, n = 3).
The activities in BCR-ABL signal pathway in leukemia cells treated by 0.25µM Givinostat were studied at various post-treatment time points and demonstrated in Western blots (Figure 1B). Obvious and significant reductions of all three key phosphoproteins in BCR-ABL signal pathways were observed in SUP-B15 beginning at 24 hours. The pBCR-ABL and pSTAT5 were virtually entirely lost at 48hr and pCrkL totally lost at 72hr. On the contrary, no inhibitory effects of Givinostat were observed on pBCR-ABL and pCrkL except pSTAT5 which were lowered at 48 and 72hrs in K562. The pCrkL protein in K562 was even slightly more at 72 hours in treated than the controls.
The inhibitory effect of Imatinib at concentrations from 0.5 to 5µM on SUP-B15 and K562 were studied (Data not shown). At 0.5µM, Imatinib inhibited about 44% (±5.0) of K562 cell growth at 48 hours, and barely inhibited SUP-B15 (16%±4.0), hence was significantly less effective than K562 (P < 0.0001) and consistent with previous finding (Quentmeier et al, 2011) [7].
Givinostat induces potent apoptosis in Pre-B ALL cells
We examined the effect of Givinostat on cell viability using both cell cycle analysis and Annexin V PI assay by flow cytometry. Givinostat at 1.0µM exhibited strongly cytotoxicity activities in SUP-B15 as evidenced by significant increases of sub-G0/G1 (apoptotic/necrotic) fractions in 24 to 48hrs (Figure 2A and Table 1). Givinostat exhibited a much less cytotoxic effect on K562. The sub-G0/G1 fractions of SUP-B15 were 37.6±5.4% and 89.9±1.9% at 24hrs and 48hrs, respectively, which were statistically significantly higher than those in K562, 18.1±3.1% and 27.8±12.8% at the same time period (P < 0.05 and P < 0.01 respectively).
Since Sub-G0/G1 fractions comprised of both apoptotic and necrotic cells, we used more accurate approach by using Annexin V-FITC/PI and quantitated by flow cytometric analysis (Figure 3) to quantify the apoptosis. With reduced Givinostat, 0.5µM, apoptosis in SUP-B15 was induced to 54% to 92.9% at the posttreatment of 24hrs to 48hrs. On the opposite hand, only 13.4% to 9.8% of K562 cells underwent apoptosis at the same doses, and differences were statistically significant when compared to SUP-B15 (P < 0.001).
The mechanisms of Givinostat-induced apoptosis in SUP-B15 were further studied by a Western blot to evaluate the status of caspase cascades. As shown in Figure 4 (SUP-B15, left panel), cleavages of caspase-3, -7 and PARP1 were detected at 24hrs and maximized at 72hrs after treatment in SUP-B15. On the contrary, except intrinsic background levels in caspase-3 and -7, all these apoptotic proteins were intact in K562 (K562, Figure 4). These data suggested that apoptosis induced by Givinostat in SUP-B15 is caspase-dependent and caspase-mediated PARP1 cleavage occurred upon caspase-activation.
The apoptosis induced by Givinostat might be p53-dependent
Apoptosis can occur via both extrinsic and intrinsic pathways and the mechanisms of Givinostat induced apoptosis in ALL cells are still unknown. In response to cellular stress, p53 mediates apoptosis through a linear pathway involving bax/cytochrome c/caspase-9 activation, followed by the activation of caspase-3, -6, and -7 cascades. Since capase-3, -7 and PARP1 activations were confirmed in Givinostat-induced apoptosis in SUP-B15 but not in K562, we postulate that p53 is functional and TP53 gene is not mutated in SUP-B15. TP53 in K562 is known in null (TP53-/-) status due to a homozygous frameshift mutation [21], and will provide an excellent cell model to confirm our postulation. Since K562 cell line has passed numerous passages since its establishment, it is critical to further confirm that K562 maintained in our laboratory still had the authentic TP53-/- status. Therefore, we reexamined the mutation status of TP53 in K562 along with SUP-B15. Using high resolution melting (HRM) technology (Materials and Methods) we first screened all of 11 exons of TP53. The mutations were then confirmed by direct DNA Sanger sequencing. In our study, the homozygous c.403_404insC in exon 5 of TP53 in K562 was confirmed by HRM and Sanger sequencing (Figure 5, right panel), which is predicted to have a nonfunctional truncate protein (p.Q135Pfsx12). In SUP-B15, only a single synonymous homozygous mutation, c.213C>G (p.P72R), in exon 4 of TP53 (Figure 5, left panel) was found.
Protein expressions of p53 and CHK1 and p21 were further analyzed by Western blot assay in cells treated with or without 0.25µM Givinostat. As shown in Figure 6 (left panel), while no significant changes in expressions of p53 and CHK1 in SUP-B15 from 24 to 72 hours, the expressions of p21, however, were elevated at 24 and 48 hours, but reduced at 72 hours as compared to untreated SUP-B15. On the contrary p53 and p21 proteins in K562 were undetectable which is consistent with the TP53-/- status. The CHK1 expression was unaffected in K562 and independent of p53/p21activities in K562.
DISCUSSION
In this study, our data confirmed that Givinostat, a Class I and II HDAC inhibitor, is a potent inhibitor for cellular proliferation and strong inducer for apoptosis in Imatinib-resistant Ph+ B-cell lymphoblastic leukemia cells, SUP-B15. This observation provides the first demonstration, to our knowledge, that apoptosis in Ph+ ALL leukemia cells induced by Givinostat requires intact p53 and is caspase-dependent.
The oncogenic protein, BCR-ABL, has a constitutively active tyrosine kinase which drives cellular proliferation and anti-apoptosis [1-3].Though Imatinib and other TKIs are potent inhibitors for CML, Ph+ B-ALL patients or established cell lines from these patients were resistant to these conventional TKIs, most likely due to high and constant expressions of phosphorylated BCR-ABL and its substrate proteins, STAT5 and CrkL, or partly due to Imatinib-induced T351I mutation in ABL domain [8. 22].In this study, in vitro treatment of lymphoblastic leukemia cells with Givinostat directly inhibited BCR-ABL signal pathway with significant loss of key phosphoproteins of pBCR-ABL, and pSTAT5 and pCrkL (Figure 1B) in a similar pattern observed in Imatinib-treated CML and K562 [11, 23],but in different mechanisms. Previous studies have shown that the HDAC inhibitor might enhance degradation of BCR-ABL proteins secondary to hyperacetylation of the chaperon protein, HSP90 [24-26].This hypothesis, however, cannot explain why inhibition on K562 by Givinostat was not observed in this study.
We studied induction of apoptosis of Givinostat on these two cell lines in hope to find the answers. Again, the apoptosis was prominently induced in SUP-B15 but not in K562. Previous study showed, though with limited numbers, that one of most important contributions to treatment failure in children and adult ALL is the presence of mutations/deletions of TP53 gene among these patients [27-28].The major anti-neoplastic function of p53 is to arrest the cells at G1 phase followed by initiating apoptosis through induction of p21 and PUMA, its transcriptional targets, as observed in IR-treated cells [29].In this study, we confirmed TP53 gene in SUP-B15 only has a homozygous p.P72R mutation, which doesn’t affect p53 expression as shown in Figure 6. First identified by Ara et al. (1990) [30],p.P72R mutation is considered as a synonymous benign mutation though Dumont et al. (2003) found that R72 had up to 15-fold increased apoptotic ability compared with P72 in both inducible Saos2 (a human osteosarcoma) cell lines and H1299 (a human lung adenocarcinoma) cells [31]. Finally p53 protein can be stabilized by the hyperacetylation of p53 [32]. Therefore, p53 might have induced p21 and its signal pathway resulting in cell cycle arrest and apoptosis in leukemia cells when the oncogenic driven factors, pBCR-ABL/pSTAT5/pCrkL, were removed by Givinostat.
On the contrary, no p53 is detectable in K562 due to the null mutation (c.403_404insC). Again these cells with defect p53 fail to neither induce p21 expression nor activate the apoptotic cascade including absent cleavage of PARP1, a sensitive marker for apoptosis at later stage.
We demonstrated in this study that in vitro treatment with a single HDAC inhibitor, Givinostat, resulted in significant inhibition on cell proliferation and induction of apoptosis in a Ph+ Pre-B ALL cell line. The anti-leukemic effect of Givinostat on Ph+ B-cell leukemia might depend on intact p53/p21. Our data strongly suggests that Givinostat can function as a potent and ideal anti-leukemic candidate drug among patients with Ph+ pre-B ALLs. A screen for TP53 mutation might be needed if it is considered to apply clinically. More studies like in vivo model to assess the therapeutic effects of Givinostat in patients-derived xenograft or clinical trials might help translate this study to clinical utilizations.
MATERIALS AND METHODS
Reagents and cell culture
Iscove’s Modified Dulbecco’s Medium (IMDM), heat-activated fetal bovine serum (FBS), and antibiotic/antifungal reagents were purchased from Life technologies (Grand Island, NY). Leukemia cell lines, K562 (AML) and SUP-B15 (ALL), were obtained from American Type Culture Collection (ATCC, Rockville, MD), and maintained in IMDM supplemented with 0.5X antibiotic/antifungal reagents and 10 to 20% FBS. The methylthiazol tetrazolium reduction (MTT) testing kit was purchased from Promega (Madison, WI). Givinostat (C24H27N3O4, 421.489 g/mol) and Imatinib (C29H31N7O, 493.603 g/mol) were purchased from Selleck Chemicals (Houston, TX), and prepared per instruction pamphlet. Antibodies used in Western blots for detection of Caspase 3 and 7, pBCR-ABL, pStat5, and pCrkL were purchased from Cell Signaling (Danvers, MA). The antibodies for detection of Checkpoint kinase 1 (CHK1), cyclin-dependent kinase inhibitor 1 (p21, or p21Cip1, or p21Waf1), tumor protein p53 (p53, DO-1), Poly (ADP-Ribose) Polymerase 1 (PARP1) and Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phosphate-buffered saline (PBS) and Radioimmunoprecipitation (RIPA) lysis buffer were purchased from Santa Cruz. The Annexin V-fluorescein isothiocyanate (FITC) kit for detection of apoptosis by flow cytometry was purchased from BD Biosciences (San Jose, CA), and Propidium Iodide (PI) from Life technologies (Grand Island, NY). Dimethyl sulfoxide (DMSO) was purchased from Sigma (St. Louis, Missouri). Cells were treated with Givinostat or Imatinib in different concentrations (0.1-1mM) for intervals (12-72hr). Cell proliferation was assessed by the MTT test as described previously [19].
Immunoblotting
Freshly cultured cells at 2.0x107 with or without treatment were harvested, washed with PBS, and re-suspended in RIPA lysis buffer containing proteinase and phosphatase inhibitors. Protein concentration was determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Sixty (60) µg of protein was separated on SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes, probed with antibodies. The blots were visualized with ECL reagent (Amersham, Arlington Heights, IL) and then exposed to autoradiography film (Denville, Metuchen, NJ). All tests were repeated three times.
Flow cytometry analysis
Freshly cultured cells at 2.0x106 with or without treatment were washed with 1xPBS, and suspended in Propidium Iodine (PI) (0.02mg/ml)/Triton X-100 (0.1%)/PBS solution with RNase A (0.2mg/ml) and stained for 1hr at room temperature (20ºC). Cells were analyzed using a flow cytometry (FACSCalliber, BD Bioscience, San Jose, CA) and data analyzed for cell cycle distribution (Mod-Fit software). For flow cytometric analysis for apoptosis using Annexin V-FITC/PI kit (BD Biosciences, San Jose, CA), 1x106 freshly cultured cells were harvested, washed with 1xPBS, re-suspend in 1x binding buffer, and then stained with 5µl of FITC/Annexin V and 5µl of PI at room temperature for 15 minutes before analyzed by flow cytometry. Fractions of viable, pre-apoptotic, apoptotic and necrosis were analyzed using FlowJo software (TreeStar).
DNA Extraction and TP53 mutation analysis
Freshly cultured cells (5x106) were harvested and washed with 1x PBS. The genomic DNA was extracted using QIAamp DNA Mini & Blood Mini Kit (Qiagen, Valencia, CA). PCR amplification was performed in a 10µl volume containing 15ng of DNA, 0.25µM of each primer (forward and reverse), 4µl 2.5X LightScanner Master Mix with LCGreen Plus Dye, and 0.5µl 100% DMSO (Sigma) and nuclease free water and followed the protocol (Idaho Technology, Salt Lake City, UT). High Resolution Melting analysis was performed on a LightScanner HR 96 (Idaho Technology). The amplicons were melted from 77°C to 96°C with a heating rate of 0.1°C per second. The data was analyzed using the LightScanner software provided by Idaho Technology. For bi-directional Sanger sequencing, the HRM amplicons were purified using QIAquick PCR Purification Kit (Qiagen, Valencia, CA) and performed as described previously [20].
Statistics
All results were expressed as means ± SD unless stated otherwise. The unpaired Student’s t test was used to evaluate the significance of differences between groups, accepting p < 0.05 as level of significance.
ACKNOWLEDGMENTS
The research was partially supported by Grant from American Cancer Society (IRG-67-003-47) and 2011 Pelotonia grant from The Ohio State University Comprehensive Cancer Center (OSUCCC) to WZ. We would like thank the administrative assistance by Shelly Belcher and Amy Glaze in the Department of Pathology, OSU Wexner Medical Center, Columbus, OH.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SPONSOR
The Ohio State University James Cancer Center Pelotonia Idea Grant (2011); American Cancer Society Grant: IRG-67-003-47.

- 1. The protooncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3’ kinase pathway. Oncogene. 1996;12:839-46. [PubMed]
- 2. Biology of chronic myelogenous leukemia-signaling pathways of initiation and transformation. Hematol Oncol Clin North Am. 2004;18:545-568, vii-viii. [PubMed]
- 3. Mechanisms of transformation by the BCR-ABL oncogene: new perspectives in the post-imatinib era. Leuk Res. 2004;28 Suppl 1:S21-28. [PubMed]
- 4. Imatinib: A targeted clinical drug development. Semin Hematol. 2003;40:15-20. [PubMed]
- 5. Results of dasatinib therapy in patients with early chronic-phase chronic myeloid leukemia. J Clin Oncol. 2010;28:398-404. [PubMed] https://doi.org/10.1200/JCO.2009.25.4920.
- 6. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med. 2001;7:228-234. [PubMed]
- 7. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J Hematol Oncol. 2011 7;4:6. [PubMed] https://doi.org/10.1186/1756-8722-4-6.
- 8. Kinase domain point mutations in Philadelphia chromosomepositive acute lymphoblastic leukemia emerge after therapy with BCR-ABL kinase inhibitors. Cancer. 2008;113:985-94. [PubMed] https://doi.org/10.1002/cncr.23666.
- 9. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108: 475-487. [PubMed]
- 10. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002; 1: 287-299. [PubMed]
- 11. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210-1216. [PubMed]
- 12. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotech 2007; 25:84-90.. 2000;92:1210-1216. [PubMed]
- 13. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18:7016-25. [PubMed]
- 14. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat). Mol Med. 2011;17:353-62. [PubMed] https://doi.org/10.2119/molmed.2011.00020.
- 15. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol. 2013;161:688-94. [PubMed]
- 16. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F). Leukemia. 2008;22:740-7. [PubMed]
- 17. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150:446-55. [PubMed]
- 18. An immediate transcriptional signature associated with response to the histone deacetylase inhibitor Givinostat in T acute lymphoblastic leukemia xenografts. Cell Death Dis. 2016 Jan 14;6:e2047. doi: 10.1038/cddis.2015.394. PubMed PMID: 26764573; PubMed Central PMCID: PMC4816177. [PubMed] https://doi.org/10.1038/cddis.2015.394.
- 19. A novel role of CYP2E1 in human megakaryocyte development. In Vivo. 2014;28:1077-84. [PubMed]
- 20. Mantle cell lymphoma 12 years after allogeneic bone marrow transplantation occurring simultaneously in recipient and donor. J Clin Oncol. 2010;28:e629-32. [PubMed]
- 21. Mutational inactivation of the p53 gene in the human erythroid leukemic K562 cell line. Leuk Res. 1993;17:104550. [PubMed]
- 22. BcrAbl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65:8912-9. [PubMed]
- 23. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of BcrAbl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63:5126-35. [PubMed]
- 24. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729-34. [PubMed]
- 25. The synthetic heat shock protein 90 (Hsp90) inhibitor EC141 induces degradation of Bcr-Abl p190 protein and apoptosis of Phpositive acute lymphoblastic leukemia cells. Invest New Drugs. 2011;29:1206-12. [PubMed] https://doi.org/10.1007/s10637-010-9465-8.
- 26. HSP90 inhibitor AUY922 induces cell death by disruption of the Bcr-Abl, Jak2 and HSP90 signaling network complex in leukemia cells. Genes Cancer. 2015;6:19-29. [PubMed] https://doi.org/10.18632/genesandcancer.49.
- 27. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185-93. [PubMed]
- 28. TP53 mutations are frequent in adult acute lymphoblastic leukemia cases negative for recurrent fusion genes and correlate with poor response to induction therapy. Haematologica. 2013;98:e59-e61. [PubMed] https://doi.org/10.3324/haematol.2012.076786.
- 29. E., Bouchier-Hayes, L., Kuwana, T., Newmeyer, D. D., Green, D. R. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732-1735. [PubMed]
- 30. Codon 72 polymorphism of the TP53 gene. Nucleic Acids Res. 1990;18:4961. [PubMed] https://doi.org/10.1093/nar/18.16.4961.
- 31. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357-65. [PubMed]
- 32. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 2003; 10:400-3. [PubMed]
Last Modified: 2016-09-28 21:30:48 EDT
 All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.