Genes & Cancer
CDK4: a master regulator of the cell cycle and its role in cancer
Stacey J. Baker1, Poulikos I. Poulikakos1,2, Hanna Y. Irie1,3, Samir Parekh2,3 and E. Premkumar Reddy1,4
1Department of Oncological Sciences, Icahn School of Medicine at Mount Sinai, Levy Place, NY 10029, USA
2Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, Levy Place, NY 10029, USA
3Department of Hematology and Medical Oncology, Icahn School of Medicine at Mount Sinai, Levy Place, NY 10029, USA
4Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, Levy Place, NY 10029, USA
Correspondence to: E. Premkumar Reddy, email: [email protected]
Keywords: CDK4/6; cancer; cell cycle; targeted therapy; checkpoint inhibitor
Received: March 10, 2022
Accepted: August 17, 2022
Published: August 25, 2022
Copyright: © 2022 Baker et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
ABSTRACT
The cell cycle is regulated in part by cyclins and their associated serine/threonine cyclin-dependent kinases, or CDKs. CDK4, in conjunction with the D-type cyclins, mediates progression through the G1 phase when the cell prepares to initiate DNA synthesis. Although Cdk4-null mutant mice are viable and cell proliferation is not significantly affected in vitro due to compensatory roles played by other CDKs, this gene plays a key role in mammalian development and cancer. This review discusses the role that CDK4 plays in cell cycle control, normal development and tumorigenesis as well as the current status and utility of approved small molecule CDK4/6 inhibitors that are currently being used as cancer therapeutics.
INTRODUCTION TO THE CELL CYCLE
The mammalian cell cycle is divided into four phases, Gap 1 (G1), Synthesis (S), Gap 2 (G2) and Mitosis (M), whose order and timing are critical for accurate transmission of genetic information. Consequently, a number of biochemical pathways have evolved to ensure that initiation of a particular cell cycle event is dependent on the accurate completion of another. These biochemical pathways have been termed “checkpoints” [reviewed in 1].
One of the major breakthroughs in our understanding of cell cycle regulation was the discovery of the cdc2+ and cdc28 genes in Schizosaccharomyces pombe and Saccharomyces cerevisiae, respectively. Both genes encode two related kinases, termed cyclin dependent kinases or CDKs, and their activities are required during the G1/S and G2/M transitions. While a single a CDK triggers the major transitions of the yeast cell division cycle, mammalian cells encode multiple CDC2-related genes [reviewed in 2]. The discovery of more than 10 different CDC2-related proteins in vertebrates initially led to speculation that regulation of the cell cycle in higher eukaryotes might involve a complex combination of CDKs and cyclins. However, subsequent studies have shown that the majority of these CDKs are not critical regulators of the cell cycle.
CDKs are holoenzymes composed of a regulatory subunit, called a cyclin, and a catalytic subunit, termed a cyclin-dependent kinase [reviewed in 1, 3, 4]. CDKs are serine/threonine kinases that have catalytic domains of roughly 300 amino acids and are inactive when underphosphorylated and monomeric [reviewed in 3]. The primary mechanism of CDK activation is the association with regulatory cyclin partners. Unlike CDKs which are highly homologous, cyclins are a remarkably diverse family of proteins, ranging in size from approximately 35–90 kDa [reviewed in 1, 3, 5]. Sequence homology amongst the cyclins tends to be concentrated in a 100 amino acid domain known as the cyclin box, which is necessary for CDK binding and activation. Complete activation of most CDKs also requires phosphorylation of a conserved threonine (Thr) residue located in the T-loop by CAK1/CDK7, a cyclin-dependent kinase that has been shown to phosphorylate the catalytic subunit of various CDKs, including the residue that is equivalent to Thr161 of CDC2, which activates the kinase activity of their holoenzymes. In CDK4, this phosphorylation occurs at Thr172 [6]. Interestingly, the activity of this protein complex does not change in a cell cycle-dependent manner and is present even in quiescent cells [7].
In mammalian cells, cell cycle progression requires a series of events that culminate in the expression and assembly of different CDKs [reviewed in 1, 3, 5]. CDK4/6 associate with D-type cyclins and mediate progression through the G1 phase when the cell prepares to initiate DNA synthesis. Activation of CDK4/6/CYCLIN D complexes contributes to hyperphosphorylation of the retinoblastoma (RB) protein and its related proteins, p107 and p130. The hypophosphorylated form of pRB binds to and sequesters several cellular proteins, and its phosphorylation results in the release of these protein factors. One key binding partner is the transcription factor E2F-1, which appears to positively activate the transcription of genes whose products are required for S-phase progression. E2F-1 and other members of the E2F family are known to bind to pRB and heterodimerize with DP-1 and -2, an interaction that is required for the DNA-binding capacity of E2F family proteins [reviewed in 1, 3–5, 8, 9]. Once the cell has made the G1/S transition, CYCLIN E/CDK2 phosphorylates the remaining residues on the RB family proteins that are critical for E2F activation. Activation of E2F-mediated transcription allows the cell to transit into S phase and initiate DNA replication, which is controlled, in part, through CYCLIN A/CDK2. CYCLIN A/CDK2 ultimately forces the cell through the G2 phase prior to the assembly of the CYCLIN B/CDK1 and the initiation of mitosis [reviewed in 9].
REGULATION OF CDK4 ACTIVITY
A key response to many growth factors in many cell types is the activation of CDK4 or CDK6 by members of the CYCLIN D family (D1, D2 and D3). Although D-type cyclins are absent in quiescent cells, they are important integrators of mitogenic signaling. CYCLIN D expression is stimulated by and is dependent on growth factors, and consequently, if these factors are removed, CYCLIN D levels drop immediately regardless of the stage of the cell cycle. A fully active CDK/CYCLIN complex can be turned off by at least two different mechanisms. Regulatory kinases can phosphorylate the CDK subunit at inhibitory sites near the N-terminus, or, CYCLIN/CDK complexes can be negatively controlled in a tissue-restricted manner by 2 families of cyclin kinase inhibitors (CKIs), the INK4 and CIP/KIP families of proteins [reviewed in 1, 10–14].
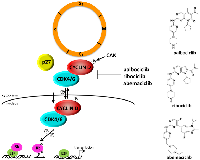
The INK4 family of proteins (p16INK4A, p15INK4B, p18INK4C, p19INK4D) inhibit D-type cyclin activity by specifically associating with CDK4 and CDK6 (Figure 1). These inhibitory proteins are expressed at low or undetectable levels in proliferating cells and are rapidly induced by growth inhibitory stimuli, such as contact inhibition, senescence, or treatment with certain growth inhibitory factors [reviewed in 11]. Of the four INK4 proteins, p16INK4A seems to play a critical role in senescence and tumor suppression in human cells [reviewed in 10, 13]. p16 is exclusively composed of four ankyrin repeat motifs, which are relatively well conserved motifs of 31-34 amino acids that mediate protein-protein interactions. In solution, the four ankyrin repeats are stacked together in a linear fashion to form a helix bundle with a concave surface which harbors clusters of charged groups that mediate protein-protein binding [reviewed in 14, 15]. The crystal structure of the p16-CDK6 complex has been solved [reviewed in 14, 15], and these studies show that binding of CDK6 to the charged domain of p16 results in an electrostatic interaction between Asp84 of p16 and Arg31 of CDK6 (which corresponds to Arg24 in CDK4). Because these residues are located in the active site of these two CDKs, this interaction diminishes kinase activity. In addition, this interaction appears to impair the binding of CDK4 and CDK6 to CYCLIN D, as it “shrinks” the CYCLIN D binding surface. This is consistent with our observation that oncogenic mutations at the Arg24 residue of CDK4 results in an inhibition of p16 binding, which in turn results in enhanced kinase activity and increased cell proliferation [16, 17].
The CIP/KIP family (p21CIP1, p27KIP1, p57KIP2) of proteins binds and inactivates CDK2/CYCLIN E, CDK2/CYCLIN A and CDK1/CYCLIN B complexes. Structure/function analysis of the p21 and p27 proteins show that their N-termini contain two key domains, one that is required for cyclin binding and another that is required for association with the CDK subunit. The cyclin binding motif appears to be important for providing high-affinity binding and is believed to underlie the specificity of CIP/KIP proteins for all cyclin-containing complexes [18, 19, reviewed in 12, 20, 21]. p27 is actually expressed throughout the cell cycle and a majority of this protein in proliferating cells is thought to be associated with CYCLIN D-CDK4 complexes. These p27-CYCLIN D-CDK4 complexes possess kinase activity, suggesting that this interaction does not result in an inhibition of CDK4 [22–30; reviewed in 11, 12]. In this case, p27 appears to stabilize CYCLIN D-CDK4 complexes, as increased expression of p27 has been shown to result in increased CDK4 kinase activity. This observation was confirmed using p27−/− mouse embryo fibroblasts (MEFs), whereby CDK4 enzymatic activity is reduced [30, 31]. Together, these studies suggest that CYCLIN D-CDK4/6 complexes exhibit a non-catalytic function, whereby their association with p21 and p27 in the G1 phase sequesters these CKIs and prevents their binding to CYCLIN E/CDK2 to allow progression through G1.
Both p21 and p27 have also been shown to inhibit the CYCLIN D/CDK4/6 complex under certain growth conditions [32–34]. p27 levels increase dramatically in response to certain anti-proliferative signals and under these conditions, cyclin D-CDK4 complexes are inactive [22, 23: reviewed in 11, 12]. These observations suggest that p27 could act both as an inhibitor and activator of CDK4-CYCLIN D complexes depending on cellular context (Figure 1). James et al. [25] reported that p27 is preferentially tyrosine phosphorylated at positions 88 and 89 in proliferating cells, causing it to bind CYCLIN D-CDK4 complexes in a non-inhibitory fashion. Treatment of tyrosine-phosphorylated p27 protein preparations with a phosphatase converted the p27 molecule to its inhibitory form, suggesting that p27 functions as an important molecular switch that discerns between growth inhibitory and growth promoting signals. The significance of the binding of p27 to CYCLIN D/CDK4 complexes and how it dictates response to targeted CDK4 therapies will be discussed later in this article.
CDK4 TARGETS
By far, the most studied G1 CYCLIN/CDK substrates is RB, which is phosphorylated in a cell cycle dependent manner. RB is hypophosphorylated in quiescent cells and becomes phosphorylated on Ser780 and Ser795 by CDK4/CDK6 during mid to late G1. The hypophosphorylated form of pRB associates with several cellular proteins, and its phosphorylation results in the disassociation of RB from its binding partners [35–37]. One such protein is the transcription factor E2F-1, which activates the transcription of genes whose products are required for S-phase progression. Most of the E2F-responsive genes identified so far are required for the G1 transition to the S phase of the cell cycle, being transcriptionally activated in a period of G1 that coincides with passage through the restriction point. The two other RB-related genes that encode pocket proteins with similar biochemical activity, p107 and p130, are also substrates of CYCLIN/CDK complexes. For example, p107 is phosphorylated by CYCLIN D/CDK4/6 on Ser842 [37]. Studies have shown that hypophosphorylated RB preferentially associates with certain histone deacetylases (HDACs) [38–40]. According to this model of RB-mediated chromatin repression, the RB-E2F1-HDAC complex binds to the promoters of S-phase specific genes, where the HDAC acts on surrounding chromatin and causes it to adopt a closed conformation. Phosphorylation of RB by CDK4/6 appears to result in the dissociation of the repressor complex, which in turn allows the expression of CYCLIN E [reviewed in 41]. CYCLIN D-CDK4/6-mediated phosphorylation of RB not only permits dissociation of the HDACs, but also appears to result in the recruitment of the CYCLIN E-CDK2 complex to the RB pocket. Under these conditions, the hypoacetylated state of chromatin is no longer maintained and histone acetylation results in the opening of chromatin structure and the activation of transcription [38–40].
In addition to these proteins, other CDK4 substrates include, but are not limited to, SMAD2/3, CDT1 and MARCKS, FOXM1, PRMT5, and several of these proteins have been shown to serve as substrates for other CDKs as well [42–46]. Interestingly, CDK4 does not phosphorylate p27 or histone H1, a canonical CDK substrate [22, 47–49], and when compared to other CDKs, the number of bona fide CDK4 substrates is relatively small [reviewed in 50]. Crystal structures of CDK4/Cyclin D complexes suggest that the active conformation of CDK4 is highly dependent on binding to both substrate and cyclin [51].
ROLE OF CDK4 IN MAMMALIAN DEVELOPMENT AND CANCER AS REVEALED BY GENETICALLY MODIFIED MOUSE MODELS
Knock-out mouse models of CDKs
As transition through each phase of the cell cycle is dependent on sequential activation of multiple CDKs, it was believed that unless there are compensatory effects by another CDK that is co-expressed (as seen with CDK4 and CDK6), loss of a single CDK would have detrimental effects on development or cell cycle progression. Such is the case of CDK1, whereby loss of CDK1 expression results in embryonic lethality at the blastocyst stage of development [52]. CDK1 is actually sufficient to drive mitosis in the absence of any interphase CDKs and can restore meiosis in oocytes, owing to its ability to bind to all cyclins and phosphorylate RB [53, 54]. Mice lacking either CDK2, 3, 4 or 6 are viable and cell proliferation is not significantly affected in vitro due to compensatory roles played by other CDKs [16, 55–59]. Nevertheless, these studies do not preclude a role for individual CDKs in mammalian development and disease. Even though Cdk2/Cdk4-null MEFs display normal S-phase progression, they eventually become immortalized and express high levels of phosphorylated RB [60]. However, knock-out of these genes in the mouse results in lethality that is likely caused by cardiac failure, a phenotype that is similar to Cyclin D1, D2 and D3 triple-knockout mice. [60, 61]. Similarly, MEFs isolated from Cdk4/6 double knock-out embryos proliferate in vitro with only slight defects in S phase, yet the embryos die in utero due to anemia [61]. Even though it was assumed that CDK4 and CDK6 have compensatory roles, knock-out of each of these loci individually has revealed unique roles for both proteins. This is not surprising given that their patterns of expression do not overlap completely. Systemic loss of Cdk6 in mice only results in a slight impairment of the mature cells that comprise the lymphoid tissues, although recent studies with conditional mouse models show a definitive role in thymocyte development, proliferation and transformation [62, 63]. The phenotype of Cdk4 null-mutant mice is actually quite different.
CDK4-knockout mice
Mice that are nullizygous for the Cdk4 allele exhibit a diabetic-like phenotype, with a 90% reduction in glucose levels, polyuria, polydipsia and dramatic reductions in the size and number of pancreatic ß-islet cells [16]. Both male and female mice are infertile, with males exhibiting testicular atrophy due to meiotic abnormalities and embryos failing to undergo implantation in females that otherwise ovulate normally [16, 64]. Females also display pituitary hypoplasia that is characterized by a reduction in the number of prolactin-producing lacotrophic cells [16, 64–67]. Interestingly, Cdk4-null animals are smaller in size compared to their wild-type littermates and it is therefore not surprising that both the size and number of both ß-islets and lacotrophs are also reduced in these animals. Although genetic rescue restores proliferation of both cell types (and thereby corrects the defects in glucose levels and female fertility) the number of these specialized cells remains reduced [68]. These data demonstrate that the observed phenotypes are the result of reduced cycling of β-cells, leading to a loss of β-islets as opposed to defects in the functionality of these cell types. That CDK4 plays a key role in homeostasis and cell cycle entry is also revealed through cell cycle experiments using serum-starved CDK4-deficient MEFs that exhibit a considerable delay in reaching S-phase and remain in G1 for prolonged periods of time [16, 17, 58]. In addition to these overt phenotypes, Cdk4 null-mutant mice are also prone to neurological defects such as impaired locomotion, staggering and hyperactivity, have abnormalities in thymocyte maturation and allergen response, and exhibit impaired adipocyte differentiation and function [16, 69, 70; reviewed in 50].
While Cdk4 null-mutant mice underscore a role for the gene in normal cell development, this animal model has also shed light on the role that this kinase plays in the genesis and progression of cancer, particularly that of the mammary gland. As discussed above, a key response to growth factors in many cell types is the activation of CDK4 or CDK6 by D-type CYCLINS. Approximately 50% of human mammary carcinomas express abnormally high levels of CYCLIN D1 [71–75] that are maintained throughout subsequent stages of breast cancer progression from in situ carcinoma to invasive carcinomas [74, 76, 77]. CDK4 is also amplified or overexpressed in a variety of tumor types, including sarcomas, gliomas, lymphomas and those of the breast [reviewed in 1].
The absence of CDK4 expression in the mammary gland results in defective mammary gland development, with the mammary glands of virgin female Cdk4-null mice displaying defects in ductal outgrowth, a reduction in the number of mammary ducts and a complete absence of alveoli [78]. Expression of the MMTV-driven Neu oncogene in wild-type animals results in the appearance of infiltrating hyperplastic and dysplastic nodules in the mammary gland that is considerably reduced in the absence of CDK4 expression. Cdk4-null females also fail to show any of the proliferative disturbances that are otherwise normally observed as a result of Neu expression. As a result, the onset and incidence of mammary carcinoma in MMTV-Neu-Cdk4−/− mice are delayed and substantially reduced, respectively. Interestingly, loss of CDK4 expression does not affect the onset or incidence of mammary tumors that result from WNT-1 expression. Although it has been reported that CYCLIN D2 expression compensates for loss of CYCLIN D1 expression in Wnt-driven mammary tumors [79], neither CDK2 nor CDK6 appear to compensate for the absence of CDK4 expression [78]. It has also been suggested that WNT and c-MYC communicate with the cell cycle machinery in breast epithelial cells through different targets during tumorigenesis in the mammary gland. In this regard, CYCLIN D1 expression is up-regulated in tumors induced by Wnt-1 and c-Myc, but not by Neu or Ras [79].
Studies on MMTV-Neu/p16 double-transgenic mice show that Neu-mediated tumorigenesis is blocked by p16 and that these double-transgenic mice develop rare tumors after a long latency [80]. Because MMTV-Neu-Cdk4−/− mice showed decreased levels of ductal branching and lobuloalveolar development of the mammary glands when compared to control animals, it is presumed that CDK4 is required for these proliferative events that are induced by Neu. These studies do not rule out the possibility that the observed defects in cdk4-null mammary gland development could be an indirect result of hormonal signaling deficiencies as opposed to an epithelial cell autonomous defect. Therefore, if the defect in mammary development observed in cdk4-null females is not cell autonomous, then WNT not only bypasses CDK4 function, but also any conceivable defects in hormone signaling resulting from Cdk4 ablation. Considering previous results indicating that Neu acts by inducing CYCLIN D1 expression, and the fact that that Cdk4 is required for Neu-induced tumorigenesis, it is probable that the CYCLIN D1/CDK4 complex itself is required for Neu-induced tumorigenesis. This is highly likely as mammary gland development in knock-in mice expressing a kinase-defective CYCLIN D1 mutant that does not associate with CDK4/6 complexes proceeds normally, and yet, these animals are resistant to Neu-induced tumorigenesis [81, 82]. However, it is important to remember that the oncogenic function of CYCLIN D1 may be partly independent of its ability to activate CDKs and is perhaps linked to the direct effects of CYCLIN D1 in controlling the expression of a subset of genes that are co-up-regulated in human tumors with deregulated CCND1 [83]. In spite of the fact that the mechanism is not fully defined, the lessons learned from Cyclin D1 and Cdk4 null mouse models have important implications with respect to therapeutic modalities that might be effective in the treatment of breast cancers that are HER2-positive.
Although cdk4-null-mutant mice highlight the importance of the CDK4/CYCLIN D1 complex in breast tumors and provide evidence to suggest that small molecule inhibitors of CDK4 kinase activity could be effective in the treatment of human disease, the importance of mutations in the CDK4 locus in human cancer was first underscored by discoveries which showed that germline mutations in this gene which abolish the ability of the encoded protein to bind to p16INK4A result in a predisposition of individuals to the development of melanoma [84, 85]. The CDK4-Arg24Cys (R24C) mutation was also detected in sporadic melanomas [84], suggesting that a CDK4 gene containing this mutation could act as a dominant oncogene that is resistant to normal physiological inhibition by p16INK4A.
CDK4R24C knock-in mice
Because the R24C mutation abolishes the ability of CDK4 to interact with p16, it was thought that the phenotype of both the cdkR24C mutant mice and the p16 null mutant mice would be identical. However, this is not the case [16, 17, 86] and their phenotype more closely resembles that of p16/p19 double knock-out animals [87]. Although CDK4R24C mice develop a variety of spontaneous primary and metastatic tumors [16, 17, 86], the major pathological abnormality observed in these animals is the onset of pancreatic islet cell hyperplasia during the first three months of life. Islets are primarily comprised of insulin-secreting ß cells and interestingly, as stated above, mice that are null for CDK4 expression develop insulin-deficient diabetes. Together, both models illustrate a critical and highly specific role for Cdk4 in the development and proliferation of this particular cell type. As expected, the CDK4R24C protein isolated from MEFs does not associate with p16INK4A and is therefore not subject to its negative regulatory effects as is evidenced by the increased expression of hyper-phosphorylated members of the RB family. CDK4R24C–expressing MEFs exhibit decreased doubling times, with a slightly higher percentage of cells in the S and G2M phases, and fail to undergo senescence. The fact that long-term cultured cells (20 passages or more) spontaneously form foci, and that the MEFs themselves are highly susceptible to Ha-ras, E1A and v-myc oncogene-driven transformation, suggests that the cdk4R24C mutation serves as a primary event in the progression towards a fully transformed phenotype [16, 17]. CDK4R24C mice are also susceptible to an increase in the development of pituitary tumors arising either in the pars intermedia or the pars distalis with characteristic angiomatous areas or dilated “blood-filled lakes” of various sizes. In many cases, the pituitary tumors compressed adjacent non-tumorous tissues, such as the hypothalamus and pons. Interestingly, mice that are heterozygous at the Rb loci and those that have disruptions in p27Kip1 and p18Ink4c also develop pituitary tumors [88–93], and it has been shown that overexpression of the high mobility group AT-hook protein 2 (HMGA2) cooperates with loss of p27 expression or expression of CDK4R24C to promote pituitary tumor development and progression [94]. While the germline R24C mutation predisposes humans to hereditary melanoma, in general, there is a low level of spontaneous melanoma occurrence in CDK4R24C mice [17, 95–97]. This observation suggests that other mutagenic events, such as exposure to UV radiation or other carcinogens could play a major role in this process, and is consistent with reports demonstrating that melanoma development in p16/p19 double knock-out mice is dependent on the expression of the H-RasG12V transgene [97].
Studies using the two-step model of skin carcinogenesis that involves sequential treatment with the mutagen 9, 10-di-methyl-1,2-benz[a]anthracene (DMBA) and tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA) and the effects on skin in Cdk4R24C mice have also been reported, but with slightly different results [86, 95–97]. We have shown that Cdk4R24C heterozygous and homozygous mice form papillomas with regions of hyperplasia in the epidermis with a very short latency period [96]. No invasion into the underlying dermis was observed and there was also a reduced incidence of benign epidermal tumors (classified as keratocanthomas), consisting of large keratin-filled cystic structures surrounded by a very well differentiated squamous epithelium [17, 96]. These results differed from what was reported by Sotillo et al. [95], who treated the animals with the same carcinogen and tumor promoter, but showed that the mice developed both papillomas and melanomas. The main differences between these two studies were the age at which the mice were treated and the dose of the DMBA/TPA. Mice that received larger doses of TPA starting at a younger age also developed melanomas. In general, treatment with DMBA/TPA results in the development of papillomas at the site of initiation and promotion with a characteristic oncogenic mutation in the 61st codon of the Ha-ras gene [98, 99] and examination of the skin papillomas in the DMBA/TPA treated mice contained this mutation. However, as only 10% of the melanomas contained mutations in the H-ras and N-ras genes in the Sotillo et al. study [95], these results suggest that other genes are targets of DMBA in these animals and/or that Ras is not necessarily the gene that “drives” melanoma initiation in the response to the R24C mutation, at least in response to DMBA/TPA.
Nevertheless, these studies do not preclude a role for Ras genes in CDK4R24C-mediated melanoma development. Several studies using tyrosinase-Hras (Tyr-HRas)/cdkR24C mice report that compound mice develop melanomas in response to DMBA/TPA and UV radiation [95–97]. These mice also develop spontaneous melanomas, albeit at a lesser frequency, and all spontaneous melanomas tested in our study showed activation of the RAS pathway [96]. While these results indicate that additional changes at the genetic level are required for maximal penetrance and tumor incidence, CDK4-deficiency in mice inhibits the development of DMBA/TPA-induced skin tumors even though the proliferation of keratinocytes and wound healing proceed normally in these animals. In normal keratinocytes, CDK6, and to a lesser extent CDK2, appear to compensate for the loss of CDK4 activity [100]. It is therefore likely that in the case of CDK4, the R24C mutation contributes to tumor progression and aggressiveness in melanomas that are initiated by H-ras activation or other changes in gene expression [101–103]. This is also likely the case with other tumor types, such as those that arise in the colons of apc+/Mincdk4R24C mice [104].
In addition to pancreatic, thyroid and skin tumors, CDK4R24C female mice develop severe mammary duct dilation and a high incidence and burden of aggressive mammary tumors [17]. The majority of the mammary tumors analyzed were adenosquamous carcinomas with papillary and cribiform elements or adenocarcinomas and adenocanthomas with squamous differentiation. The tumor cells lining the cavities undergo squamous differentiation or metaplasia with keratinization and formation of laminated horny pearls. This is consistent with the observation that MMTV-cyclin D1 transgenic mice are prone to a high frequency of adenocarcinomas and adenocanthomas [105]. As Cdk4 is required for vHa-ras-driven mammary tumorigenesis [106], and given that CDK4R24C females have an increased incidence of spontaneous mammary tumors and that the CDK4R24C protein cooperates with RAS to drive melanoma formation, it is surprising that co-expression of CDK4R24C and vHa-ras in mammary epithelial cells delays the onset of tumorigenesis. This is not due to reduced proliferation as both control and CDK4R24C-expressing tumors express comparable levels of proliferative markers. Rather, expression of RAS and CDK4R24C leads to activation of senescence pathways and induction of apoptotic and DNA damage pathways [106]. Loss of CDK4 expression also results in the senescence of pre-neoplastic cells in the lung and blocks the development of lung tumors in mouse models [107]. Although the KRAS-G12V transgene is expressed in multiple tissues, tumor induction and subsequent senescence due to an absence of CDK4 expression were only detected in the lung. It is at present unclear as to why only cells of the lung are dramatically affected. These observations are consistent with the RAS isoform and codon mutation biases that are typically present in malignancies of the lung [reviewed in 108], although it is possible that there are additional changes in gene expression that occur only in lung tumor tissue. Given that oncogenic mutations can result in a tumor cell’s dependence on CDK4, and that CDK4 regulates breast cancer tumor cell stemness [109], these studies suggest that the development of CDK4-specific inhibitors may be beneficial in the treatment of cancer types that rely predominantly on CDK4 expression.
CDK4 AND ITS ROLE IN NORMAL AND TUMOR CELL METABOLISM
While cell cycle regulatory proteins such as Rb, CYCLIN D3 and CDK9, regulate lipid and oxidative metabolism in adipocytes [110–112, reviewed in 113], the possibility that CDK4 itself might play a role in metabolism arose from the observations that Cdk4-null mutant mice develop a diabetic-like phenotype [22] and that disruption Cdk4 alleles or expression of those that encode the R24C activating mutations in primary MEFs results in reduced and increased adipogenic potential, respectively [69]. Furthermore, adipocytes isolated from Cdk4−/− mice exhibit decreased expression of genes that play a role in lipogenesis, decreased insulin sensitivity and glucose uptake [69]. CDK4 has since been shown to be an essential mediator of insulin response in adipocytes [114].
Studies investigating the role that CDK4 plays in glucose metabolism revealed that in mice, the role that CDK4 plays in glucose metabolism is independent of its cell cycle regulatory activities. Insulin activates CYCLIN D1-CDK4 complexes which suppress glucose production in the liver via the GCN5 (general control non-repressed protein 5) histone acetyltransferase [115]. Amino acids in the diet actually increase the level of Ccnd1 mRNA and CYCLIND1/CDK4 complexes activate GCN5, resulting in an inhibition of gene expression of genes that regulate gluconeogenesis. In agreement with these studies, levels of CYCLIND1/CDK4 complexes are increased in mouse models of diabetes and remain at an increased steady state level, even in response to changes in food intake and/or fasting [115].
In addition to glucose metabolism, CDK4 and CYCLIN D3 also plays a role anaerobic glycolysis and fatty acid oxidation (FAO) via modulation of AMPK (AMP-activated protein kinase) activity [116]. In this setting, CDK4 negatively regulates FAO via phosphorylation of the AMPKα2 subunit. While Cdk4 null mutant MEFs phenocopy cells that have been treated with an AMPK activator and display high levels of FAO and low levels anaerobic glycolytic activity, cells expressing the activating CDK4R24C mutant protein exhibit the opposite phenotypes. Because metabolic pathways are often subverted in tumor cells, the roles that CDK4 and CDK6 play in malignant cell metabolism as it relates to cell survival is an active area of research. For example, studies have shown that in tumors that express high levels of CYCLIN D3/CDK6 complexes, they simultaneously regulate cell cycle progression and survival, in addition to directring a metabolic shift to the the pentose phosphate shunt. Interestingly, tumor types that express other forms of CYCLIN D-CDK4/6 complexes, such as breast cancers in which CDK4 is often in complex with CYCLIN D1, do not undergo apoptosis or exhibit changes in the phosphorylation status of metabolic enzynes in response to CDK4 inhibition [117]. The role that CDK4 plays in cancer and the development and utility of CDK4/6-targeted therapies in various tumor types are discussed in greater detail in the remainder of this article.
THE ROLE OF CDK4 IN HUMAN CANCER
It is now believed that a vast number of human tumors exhibit deregulation of the CDK4/6-CYCLIN D-INK4-RB pathway by multiple mechanisms. For example, CDK4/6 is hyperactivated in many human cancers as a result of overexpression of positive regulators such as CYCLIN D, inactivation of INK4 and CIP/KIP inhibitors, or deletion and/or epigenetic alterations of substrates such as RB [reviewed in 1, 118, 119]. Hyperactive CDK4 has been reported in epithelial malignancies in the endocrine tissues and mucosa while CDK6 activation was reported in certain mesenchymal tumors such as sarcomas and leukemias [reviewed in 1]. Mutations and chromosomal translocations in the CDK4 and CDK6 loci have also been described. One of the best examples is the CDK4R24C mutation that results in insensitivity to INK4 family inhibitors and was first described in patients with familial melanoma [84, 85]. An analogous point mutation in Cdk6 that blocks the interaction of p16INK4a with CDK6 has also been reported in a human neuroblastoma cell line [120]. Chromosomal translocations within the CDK6 promoter that lead to CDK6 overexpression were also described in splenic marginal zone lymphomas and B-cell lymphocytic leukemias [121, 122]. Finally, CDK4/6 amplification or overexpression has also been observed in a wide spectrum of tumors, including gliomas, sarcomas, lymphomas, melanomas, cancers of the breast, squamous cell carcinomas and leukemias [reviewed in 118, 123].
CDK4 expression is also a prognostic indicator in certain cancers, such as triple negative breast cancer (TNBC). Studies have shown that CDK4 is highly expressed in this tumor type and correlates with poor survival and gene signatures that are associated with metastasis [109]. One of the primary reasons for TNBC recurrence is thought to be due to the presence of disproportionately large numbers breast cancer stem cells (BCSCs) within these tumors. These cells are a sub-population of cells which are resistant to standard chemotherapeutic agents, are long-lived, have the ability to self-renew, grow as mammospheres in vitro and initiate tumor development in mouse models [124–127]. CDK4 is a regulator of TNBC BCSC self-renewal and does so, in part, by down-regulating the expression of bone morphogenic protein-4 (BMP-4) [109], a key player in tumor stem cell biology [128]. Importantly, CDK4 also promotes the self-renewal and proliferation of chemotherapy-resistant TNBC BCSCs, which are largely responsible for the recurrence and metastasis in this aggressive cancer [109]. Given that inhibition of CDK4 activity in TNBC cell lines results in BCSC differentiation [129] and loss of self-renewal [109], blocking CDK4 activity in this and other tumor types is a sound approach and has resulted in the approval of several CDK4/6 ATP-mimetics for use in the clinic.
TARGETING CDK4 FOR CANCER THERAPY
Approved small molecule inhibitors of CDK4/6
Early evidence that CYCLIN D and CDK4/6 activities are upregulated in certain tumor cell types, and that cdk4-/- mice fail to develop MMTV-neu and MMTV-ras-induced mammary tumors, led to concerted efforts to develop small molecule inhibitors for these kinases. The first generation of CDK inhibitors developed, e.g., flavopiridol and roscovitine, potently inhibited CDK4 but were non-selective, inhibited multiple kinases and caused severe toxic side effects when these molecules entered clinical trials [reviewed in 21, 130].
In an attempt to overcome the toxicity profile of pan-CDK inhibitors, several groups undertook the initial development of next generation CDK inhibitors that are specific for individual CDKs. Some of these compounds exhibited a high degree of selectivity towards CDK4/6 by targeting the ATP binding site of CDK4/6-CYCLIN D complexes. Structures and IC50 values of potent CDK4/6 selective compounds, members of chemical classes of oxindoles, triaminopyrimidines, diarylureas, thioacridones, benzothiadiazines, indolocarbazoles, and pyrido[2,3-d]pyrimidines, have been summarized elsewhere [reviewed in 130, 131]. Of these, one CDK4/6 selective compound, palbociclib (PD-0332991, Ibrance®) (Figure 1), a pyrido[2,3-d]pyrimidine derivative, is exquisitely specific for CDK4 and CDK6, and inhibits these two kinases in vitro with IC50 values of 0.011 and 0.015µmol/L, respectively [132]. In its early stages of pre-clinical development, this compound was extensively studied for its efficacy in RB-positive tissue culture model systems as well as in mouse xenograft models of colorectal cancer (CRC), mantle cell lymphoma (MCL) and disseminated myeloma, where it induced G1 arrest and showed excellent efficacy [132–141]. Therapeutic doses of palbociclib resulted in a marked reduction of both phosphorylated RB and the proliferative marker Ki-67 in the tumor tissue and the downregulation of E2F-target genes. Based on these very promising results, this compound entered Phase I clinical trials in 2004, with early results indicating tolerable side effects [133–136, 142–144; reviewed in 50]. Unfortunately, the efficacy profile of this compound as a single agent was somewhat disappointing, resulting in disease stabilization rather than regression. However, results from phase II and III trials testing palbociclib in combination therapy were far more encouraging. Accelerated approval for palbociclib in combination with letrozole (Femara®), an aromatase inhibitor, for the treatment of advanced ER+HER2- breast cancer was granted in 2015 based on the results of the Phase II PALOMA-1 trial. Improved progression-free survival was observed with palbociclib and letrozole combination therapy compared to treatment with letrozole as a single agent in the subsequent Phase III PALOMA-2 trial. Finally, approval of palbociclib for a second indication, in combination with fulvestrant following progression on endocrine therapy, was granted based on the phase 3 PALOMA-3 study [145, 146]. Today, palbociclib is widely used in the treatment of advanced hormone receptor-positive (HR+) metastatic breast cancer (MBC) in combination with either an aromatase inhibitor or fulvestrant [reviewed in 147–149].
Owing to the success of palbociclib, additional orally bio-available CDK4 inhibitors with low nanomolar IC50 values against CDK4/6 have been approved for use in the treatment of HR+/HER2- metastatic breast cancers (MBCs) in combination with other approved therapies. Ribocilib (LEE011, Kisqali®) (Figure 1) received approval from the FDA in 2017 and inhibits CDK4 and CDK6 with IC50 values of 10 and 39 nM, respectively [150, 151; reviewed in 152–154]. Although effective as a single agent, like palbociclib, combination of ribociclib with an anti-estrogen is more effective in inhibiting tumor growth and RB phosphorylation [151]. Similarly, abemaciclib (LY2835219, Verzenio®) (Figure 1), which inhibits CDK4 and CDK6 with IC50 values of 2 and 5 nM, respectively, has received approval from the FDA for use in HR+/HER2- MBCs as a monotherapy for patients with progressive disease that are being treated with endocrine and chemotherapy and in combination with fulvestrant for patients whose disease has progressed following endocrine therapy [reviewed in 153].
Given the survival benefits for advanced ER+ breast cancer observed with all of the CDK4/6 inhibitors developed thus far, their use in adjuvant therapy for early stage, high-risk ER+ breast cancer was studied in several subsequent clinical trials, with somewhat conflicting results. Although a benefit with respect to improved disease-free survival was observed in the MonarchE trial [155] favoring the addition of abemaciclib for two years to standard endocrine therapy, there was no significant survival difference with the addition of palbociclib to endocrine therapy in the PALLAS study [156]. While there were slight differences in the enrolled patient populations, there may be intrinsic biological differences among the different CDK4/6 inhibitors.
The utility of CDK4/6 inhibitors as monotherapy or in combination with other targeted therapeutics is being explored in breast cancer and other tumor types. Agents that block PI3K signaling are of particular interest as the latter is activated in greater than 70% of breast cancers [157]. Pre-clinical studies using TNBC patient derived xenograft and immunocompetent syngeneic animal models have shown that combining CDK4/6 inhibitors with those that block CK1ε [158] as well PI3Kα [159] resulted cell cycle arrest, apoptosis, the activation of tumor infiltrating T-cells and an increase tumor immunogenicity. Short-term inhibition of CDK4/6 has also been shown to sensitize ER+ breast cancers to radiation therapy in pre-clinical models [160], highlighting an additional use for these agents in combination therapy.
BIOMARKERS OF CDK4/6 INHIBITOR RESPONSE
In preclinical studies using breast cancer cell lines, an intact RB axis was required for sensitivity to CDK4/6 inhibitors. These cell lines and tumors, which express functional RB, responded remarkably well to combination therapy using CDK4/6 inhibitors with ER antagonists when compared to use of either inhibitor as a single agent as evidenced by loss of RB phosphorylation, a reliable biomarker of response to CDK4. However, functional RB may not be a requirement for response in other pre-clinical cancer models [reviewed in 147, 148]. Given the rarity of RB gene deletion/mutation in ER+ breast cancer, multiple genes or gene signatures may be required to accurately predict clinical sensitivity to these inhibitors. Several candidates such as CCND1 (amplification) and p16INK4A (loss) have been studied as single biomarkers of sensitivity but have not borne out in the PALOMA-1 trial and other clinical studies. Consequently signatures aimed at identifying tumors with CYCLIN D activation, RB loss of function or CDK4 inactivation are being explored in clinical samples to distinguish those cancers that are sensitive or resistant to CDK4/6 inhibitor therapies [reviewed in 161]. For example, in the NeoRHEA Phase II trial, biopsies obtained before and after neoadjuvant treatment with palbociclib and endocrine therapy are being used to validate a CDK4 inactivation gene signature as a biomarker of insensitivity to CDK4/6 inhibition [162].
Toxicities associated with CDK4/6 inhibitors
The common side effects of CDK4/6 inhibitors can be classified into hematological and non-hematological toxicities. Although all act by a similar mechanism, the side effect profiles are different for each agent, with neutropenia and leukopenia more frequently associated with palbociclib and ribociclib whereas diarrhea is more frequently seen with abemaciclib.
Hematological toxicities
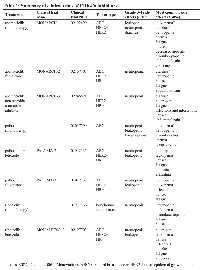
Myelosuppression has emerged as a class effect of CDK4/6 inhibitors. Even though grade 3 and 4 neutropenias are very common in the published trials, the rate of infections was less frequent (25–58%) and febrile neutropenia did not exceed 2%. Moreover, the neutropenia is completely reversible without G-CSF within seven days of treatment withdrawal. This suggests that the myelosuppression from CDK4/6 inhibition is different from conventional chemotherapy-induced neutropenia. The mechanism that underlies this observation has been demonstrated in vitro to be due to transient cell cycle arrest in bone marrow progenitors [163]. Hence, the dosing schedule recommended for the inhibitors is cyclical, for three weeks on with one week off. Myelosuppression usually emerges 15 days after the first dose of palbociclib and ribociclib, and with abemaciclib, it emerges within the first two cycles and then less frequently in subsequent cycles. Anemia and thrombocytopenia are also commonly seen in patients treated with CDK4 inhibitors (Table 1) without increases in bleeding or transfusion requirements. Treatment guidelines have been established for blood count monitoring and dose reduction due to cytopenias for this class of drugs [164–166].
Non-hematological toxicities
Common non-hematological toxicities associated with CDK4/6 inhibitors include fatigue, diarrhea, transaminitis, increased creatinine and Qt prolongation. Diarrhea, abdominal pain and increased creatinine were seen more frequently in clinical trials with abemaciclib. In addition to blood count monitoring, liver function and Qt assessments at baseline are recommended with these drugs. More recently, the FDA has warned that rare but severe interstitial pneumonitis can be observed with CDK4/6 inhibitor treatments [167].
CDK4 inhibitors as modulators of immune checkpoints
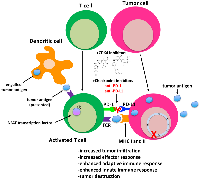
The role that the immune system plays in controlling tumor growth and how tumors evade the immune system has recently been thrust into the limelight as the utility of checkpoint inhibitors in cancer therapy continues to rise. While the main function of immune checkpoints is to maintain self-tolerance [168], it is now well established that cancer cells co-opt these pathways to evade destruction by the immune system. In certain cancers, such as those of the lung, kidney and skin (melanoma), immunotherapy is considered standard of care for subsets of patients with these particular tumor types [169]. However, in breast cancer and other tumor types, overall response to checkpoint inhibitors is relatively low (<20%) when administered as single agents [170]. Interestingly, it has recently been shown that CDK4/6 inhibitors induce an anti-tumor immunogenic response by 4 primary mechanisms: (1) increased antigen presentation in the tumor itself via innate and adaptive immunity, (2) enhanced T-cell activation, (3) suppressed proliferation of immunoresponsive regulatory T cells (Tregs) and (4) induction of a memory T cell phenotype [159, 171–176]. In an effort to expand the utility and benefits of immune checkpoint therapy to breast and other cancers, several groups have exploited the immunomodulatory properties of CDK4/6 inhibitors to enhance the efficacy of immune checkpoint inhibitors. Studies in animal models clearly demonstrate that abemaciclib and palbociclib synergize with anti-PD-1 and PD-L1 antibodies to induce tumor regression, and in some cases complete regression, in immunocompetent mouse models of lung [172] and mammary tumors [159, 160, 173, 174].
Several mechanisms appear to underlie the synergism between checkpoint and CDK4/6 inhibitors (Figure 2). PD-L1 expression fluctuates during the cell cycle, with maximum levels observed in M phase and declining sharply thereafter [173]. Disruption of ccnd1-3 loci and that of cdk4 (but not cdk6) in MEFs, as well as treatment of tumor cell lines with CDK4 inhibitors, also stabilize PD-L1 at the protein level [173]. Inhibition of CDK4 also blocks its phosphorylation of speckle-type POZ protein (SPOP), a CULLIN-3 E3 ligase, and leads to the degradation of SPOP by FZR1, a component of the anaphase promoting complex. Because CULLIN-3SPOP interacts with the PD-L1 cytoplasmic domain, PD-L1 expression is stabilized in tumors treated with CDK4 inhibitors [172]. Treatment of tumor bearing mice with CDK4/6 inhibitors has a priming effect on CD8+ T cells, leading to a memory phenotype that has a distinct gene signature and is RB-dependent [175, 176]. CDK4/6 inhibition also has a direct effect on T cell receptor (TCR) signaling by triggering nuclear localization of the nuclear factor of activated T cells (NFAT) transcription and NFAT-dependent changes in gene expression that are essential for T cell activation [172, 174]. At the genomic level, CDK4 inhibitors induce changes in the expression of genes that are associated with a T-cell-mediated inflammatory response, dendritic cell maturation and antigen presentation via downregulation of DNMT1 [171, 174]. Accordingly, analysis of tumors treated with these inhibitors show upregulation of major histocompatibility (MHC) class I and II and antigen presenting cells [174].
In spite of these observations, early phase I/II clinical data on combination CDK4/6 inhibitor/letrozole and anti-PD-1 (pembrolizumab) treatment demonstrated increased but tolerable toxicities in the absence of improved efficacy in terms of survival [177] when compared to that observed in the earlier PALOMA-2 clinical trial. Mechanistically, no significant differences in tumor-infiltrating lymphocytes, PD-L1 levels, or gene-expression profiles that are typically associated with CDK4/6 inhibitor resistance were observed between patients who showed a complete response versus those who did not [177]. As these data represent some of the earliest trial results obtained for CDK4/6 inhibitor/immunotherapy combination treatment, additional studies will determine whether similar treatment regimens are of clinical benefit.
Paradoxically, inhibiting CDK4/6 might be a double-edged sword. Studies using a genetically modified mouse model of Eµ-myc-induced B-cell lymphoma showed that disruption of the cdk4 locus leads to genomic instability and accelerated lymphoma development via FOXO1 [178]. Analysis of human non-Hodgkin B-cell lymphoma specimens showed that CDK4 protein expression is down-regulated in several sub-types, which correlated with reduced levels of CDK4 transcripts, pointing to a tumor-suppressive role for CDK4 in MYC-driven B-lineage malignancies via suppression of genomic instability. These conclusions are supported by previous studies of CDK6, which is also targeted by palbociclib and other CDK4 small molecule inhibitors, and its role in BCR-ABL transformed B lineage cells. Kolmann et al. [179] demonstrated that overexpression of CDK6 in such cells delayed their cell cycle progression and suppressed their ability to promote leukemogenesis in mice by inducing expression of p16INK4A. The authors also reported that expression of CDK6 inversely correlated with that of p16INK4A in human B- and T-cell lymphomas. Together, these and other studies in lymphoid cells suggest that therapeutic targeting of CDK4/6 might promote unwanted phenotypes in a cell-type dependent manner.
Resistance to small molecule CDK4/6 inhibitors
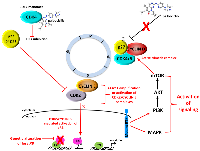
There are several mechanisms by which tumors can acquire resistance to or simply fail to respond to CDK4/6 inhibition [reviewed in 180] (Figure 3). As previously mentioned, the earliest studies that pre-dated clinical approval of these agents in ER+ MBC demonstrated that response was largely observed in ER+ vs. ER- breast cancers, suggesting that mutations in estrogen receptor (ESR) genes and others that play a role in ER signaling might lead to resistance. Although mutations in the ESR1 gene can be responsible for resistance to endocrine therapy [181–184], they do not drive resistance to CDK4/6 inhibition [185]. A recent study of MBCs with acquired resistance to ER targeted therapies revealed that mutations in ERBB2 not only conferred estrogen-independent growth and resistance to fulvestrant and tamoxifen, but also resistance to palbociclib, suggesting that use of a HER2 inhibitor would be effective in this setting [186]. Given that an intact RB pathway is key to response, a second and more obvious cause of resistance is loss or mutation of RB as well as other genes encoding cell cycle machinery. While not an all-inclusive list, studies using established breast tumor cell line and patient-derived xenograft models as well as retrospective analysis of clinical trial data have shown that resistance to CDK4/6 inhibition is associated with downregulation of RB expression, activation of interferon (IFN) signaling, FGFR1 amplification, overexpression of CCNE1 and overexpression/activation of CDK2/CYCLIN E1 complexes [187–189, reviewed in 190, 191].
Cells that are resistant to CDK4/6 inhibition also employ alternative mechanisms to drive proliferation and escape growth arrest [reviewed in 191, 192]. One of the initial kinome-wide studies aimed at identifying druggable pathways in CDK4/6 inhibitor-resistant cells revealed that 3-phosphoinositide-dependent kinase 1 (PDK1) was upregulated in terms of overall expression levels and phosphorylation in cells treated that acquired resistance to ribociclib [193]. Consistent with previous studies, cell cycle progression in these cell lines was driven by S-phase CYCLIN/CDK2 complexes. Both genetic and pharmacologic inhibition of PDK1 sensitized ER+ breast cancer cells to and acted synergistically with ribociclib in vitro and in vivo. Combination studies also revealed that cells treated with these inhibitors underwent apoptosis, which is in contrast to the cytostatic effects and senescence-inducing properties of CDK4/6 inhibitors [193]. The results of subsequent RNA-Seq and kinome analyses have continued to underscore reliance on the phosphatidyl-inositol-3-kinase/mammalian target of rapamycin (PI3K-mTOR) signaling axis [194] in CDK4/6 inhibitor-resistant cells, whereby mTORC1/2 inhibition in palbociclib-resistant cells reduces tumor growth [195, 196] and reactivates the CDK4/6-RB signaling node with commensurate changes in E2F-mediated transcription [195], without inducing a senescence-like phenotype. The fact that abnormal PI3K-mTOR as well as MAPK signaling are observed in multiple tumor types that exhibit do novo or acquired resistance to CDK4/6 inhibitors [reviewed in 191, 192] suggests that targeting these pathways might be of significant clinical value. A number of clinical trials with PI3K/mTOR-targeted therapies in combination with CDK4/6 inhibitors for use in advanced HER2+/− breast cancer are ongoing [reviewed in 191], as those that are biomarker-driven in order to assess what agents would be beneficial to patients as second-line therapeutic regimens. Phase I-III trials examining the therapeutic benefit of selective estrogen receptor degraders as well as small molecule CDK2, BCL-2 (venetoclax/Venclexta®), FGFR, AURKA and immune checkpoint inhibitors (discussed above) are also active and/or completed [reviewed in 197].
Mechanism of action of CDK4/6 inhibitors
X-ray crystallography studies have revealed a surprising mechanism of action associated with palbociclib-mediated inhibition of CDK4 [198]. Dr. Rubin’s group determined the crystal structure of the CDK4 holoenzyme, which revealed that p27 binds to CDK4 and allosterically activates it by remodeling the ATP-binding site, thereby promoting release of the kinase activation segment leading to RB phosphorylation. They also found that phosphorylation of Tyr74 in p27 is essential for the activation CDK4 and that the lack of such a tyrosine residue in p21 makes it a poor activator of CDK4. As previously discussed, p27 is expressed throughout the cell cycle where it is associated with CYCLIN D-CDK4 complexes [22, 23, 25–29; reviewed in 11, 12]. However, the mechanism by which it activates these complexes was not well defined. In this context, studies by Guiley et al. [198] show that the CYCLIN D1-CDK4-p27 trimeric complex is resistant to the effects palbociclib and other approved CDK4 inhibitors. Instead, palbociclib primarily targets CDK4/6 monomer and promotes the formation of inactive CDK2-p21 complexes leading to inactivation of CDK2 enzyme activity, which is essential for sustained RB inhibition and growth arrest [198] (Figure 3). Subsequent stability studies by Pack et al. [199] using the 3 clinically approved CDK4/6 inhibitors have shown that they induce immediate dissociation of p21 from CDK4 while leaving CDK6 complexes intact. Therefore, while p21 is able to indirectly inhibit CDK2, p27 is unable to do so due to the fact that it remains bound to CDK4/CYCLIN D1 complexes [199] which are refractory to CDK4 inhibitors. As the authors of both studies study point out, inhibition of CDK2 is of paramount importance for the induction of cell cycle arrest in breast tumor cells that respond to CDK4/6 inhibitors and without CDK2 inhibition, patients will undoubtedly develop resistance to this class of therapeutics. Ongoing and future clinical trials examining the utility of CDK2 inhibitors in patients that progress while undergoing CDK4/6 inhibitor treatment will hopefully address this issue.
Despite the effectiveness of CDK4/6 inhibitors in HR+ breast cancer, their preclinical and clinical effectiveness has been limited in other predominantly RB-proficient tumor types, such as in NSCLC, colorectal cancer and melanoma [200, 201]. Closer examination revealed that expression of CDK6 affects response to CDK4/6 inhibitors. Tumors with low levels of CDK6 compared to CDK4 are universally sensitive to CDK4/6 inhibitors, including HR+ breast, mantle cell lymphomas, Ewing sarcomas as well as subsets of large tumor types, e.g. NSCLC. By contrast, when both CDK6 and CDK4 are coexpressed in a tumor at comparable levels, CDK6 drives Rb/E2F output. In the vast majority of RB-proficient solid tumors that are intrinsically resistant to CDK4/6 inhibitors, CDK6 is expressed in a thermostable conformation (CDK6-stable) with weak binding to current CDK4/6 inhibitors [202]. New inhibitors able to bind and inhibit CDK6-stable are warranted to effectively suppress Rb/E2F output in these tumors.
CDK4/6 PROTACs
The strategy of Proteolysis Targeted Chimeras (PROTACs) has recently gained traction in the field of drug development [reviewed in 203]. PROTACs are heterobifunctional small molecules that include a ligand binding to a target protein and a moiety binding to an E3 ubiquitin ligase, conjugated by a linker aimed at degrading the target protein through the ubiquitin-proteasome system. A number of recent studies have reported on CDK4/6-directed PROTACs and their potential use as biochemical tools or potential therapeutics [204–210]. A notable sub-class of PROTACs showed significantly better degradation of CDK6 over CDK4, suggesting a potential therapeutic use in certain leukemias such as in AML and Ph+ ALL [202, 210, 211]. CDK4/6-directed PROTACs can be also used as a tool to indirectly determine target engagement in living cells. In one study, a CDK4/6 directed PROTAC revealed weak target engagement of CDK6 by CDK4/6 inhibitor in many solid tumors, indicating intrinsic resistance to CDK4/6 inhibitors. In the same solid tumor cells, a CDK6-selective PROTAC was also unable to degrade CDK6 [202], emphasizing the need for development of potent inhibitors of CDK6-stable to be used in future PROTAC development for these tumors.
Another distinguishing feature across CDK4/6 PROTACs that may potentially influence their therapeutic implications, is the ability of some but not all to induce degradation of the canonical “imid” targets, Icaros and Aiolos. Other studies are needed to determine whether this additional property will contribute increased antitumor effectiveness, as suggested by Jiang et al. [205], or if it will potentially add toxicities.
A NON-THERAPEUTIC ROLE FOR CDK4 INHIBITORS AS PREVENTATIVE AGENTS FOR CHEMOTHERAPY-INDUCED DEPLETION OF HEMATOPOIETIC STEM CELLS
One of the unfortunate side effects of systemic chemotherapy with traditional cytotoxic agents is immunosuppression that arises due to bone marrow (BM) toxicity, with the acute effects being predominantly due to the destruction of hematopoietic stem cells and progenitors (HSPCs). The proliferation of these cell types is dependent on CDK4/6 activity due to the fact that CYCLIND1/CDK4 complexes drive the G0-G1 transition and shorten the length of the G1 phase [212]. In a recent study, He et al. [213] reported that treatment of mice with trilaciclib (G1T28), a clinically-approved CDK4/6 inhibitor that was developed to reduce chemotherapy-induced myelosuppression [214], transiently arrests HSCs and progenitors in the G1 phase of the cell cycle. Although this result was not surprising, the authors cleverly extended these studies to show that a single dose of trilaciclib 30 minutes prior to a single or repeated dose of 5-fluorouracil (5-FU) also induces a transient G1 arrest in the hematopoietic stem and progenitor cells (HSPCs) in mice, preserved HSC function and reduced the degree of myelosuppression due to HSC exhaustion. These effects were intrinsic to the BM and lasted long-term as demonstrated by BM transplantation assays. Interestingly, primary and secondary transplant recipients did not develop any hematological malignancies in a 1-year time period following repeated combination dosing, suggesting that trilaciclib and other CDK4/6 inhibitors might also prevent secondary myeloid neoplasms that emerge in a small percentage of patients treated with systemic chemotherapeutics that induce HSC exhaustion and myelosuppression. Further studies using syngeneic or other immunocompetent models with tumors that are not dependent on CDK4/6 activity, as well as determining whether the non-hematological AEs associated with CDK4/6 inhibitors (discussed above) are likely to shed further light on the utility of these drugs in the context of HSPC protection.
SUMMARY
CDK4/6 is a key mediator of cell cycle progression through the G1 phase, the time when a cell prepares to initiate DNA synthesis. The cell’s reliance on this protein as well as its CYCLIN D binding partners and downstream target RB for proliferation underscores why the CDK4/CYCLIN D/RB signaling module is often deregulated in transformed cells. The approval of 3 CDK4/6 inhibitors as treatments for ER+ breast cancer has paved the way for ongoing clinical studies evaluating the utility of these inhibitors in combination with those of other signaling pathways (such as but not limited to BRAF, PI3K and MEK) in multiple tumor types that exhibit reliance on CYCLIN D1/CDK4/RB or other components of the cell cycle such as p16 and p27. The success of these trials, as well as understanding the mechanisms that drive resistance to these inhibitors, should provide an answer as to whether selective inhibitors of CDK4/6 can provide therapeutic benefit in a broader array of cancers.
Abbreviations
BCSC: breast cancer stem cell; BM: bone marrow; CDK: cyclin-dependent kinase; ER: estrogen receptor; HR: hormone receptor; HSC: hematopoietic stem cell; MBC: metastatic breast cancer; MMTV: mouse mammary tumor virus; PD-1: programmed cell death protein; PD-L1: programmed death-ligand 1; PROTAC: proteolysis targeting chimera; RB: retinoblastoma.
Author contributions
SJB, SP, HYI, PIP and EPR wrote the manuscript.
ACKNOWLEDGMENTS
P.I.P is supported by grants from the National Institutes of Health (NIH) and National Cancer Institute (NCI) (R01 CA204314, R01 CA240362, and R01 CA238229), the Irma T. Hirschl Trust, the Manhasset Women’s Coalition Against Breast Cancer, the Breast Cancer Alliance, the Melanoma Research Foundation, the Melanoma Research Alliance and Tisch Cancer Institute (TCI) developmental awards. H.Y.I. is supported by grants from the American Cancer Society (ACS) and the Breast Cancer Research Foundation (BCRF). S.P. is supported by the NCI (R01CA244899 and R01 CA252222) and receives research funding from Amgen, Bristol Myers Squibb and Karyopharm. E.P.R is supported by a grant from the NIH/NCI (RO1 CA251448).
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
- 1. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009; 9:153–66. https://doi.org/10.1038/nrc2602. [PubMed]
- 2. The yeast cell cycle. Curr Opin Cell Biol. 1991; 3:242–46. https://doi.org/10.1016/0955-0674(91)90147-q. [PubMed]
- 3. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997; 13:261–91. https://doi.org/10.1146/annurev.cellbio.13.1.261. [PubMed]
- 4. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001; 1:222–31. https://doi.org/10.1038/35106065. [PubMed]
- 5. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene. 1995; 11:211–19. [PubMed]
- 6. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol Cell Biol. 1994; 14:2713–21. https://doi.org/10.1128/mcb.14.4.2713-2721.1994. [PubMed]
- 7. Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes Dev. 1998; 12:370–81. https://doi.org/10.1101/gad.12.3.370. [PubMed]
- 8. The regulation of E2F by pRB-family proteins. Genes Dev. 1998; 12:2245–62. https://doi.org/10.1101/gad.12.15.2245. [PubMed]
- 9. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000; 14:2393–409. https://doi.org/10.1101/gad.813200. [PubMed]
- 10. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006; 7:667–77. https://doi.org/10.1038/nrm1987. [PubMed]
- 11. Switching cyclin D-Cdk4 kinase activity on and off. Cell Cycle. 2008; 7:892–98. https://doi.org/10.4161/cc.7.7.5637. [PubMed]
- 12. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004; 18:851–55. https://doi.org/10.1101/gad.1205304. [PubMed]
- 13. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life. 2007; 59:419–26. https://doi.org/10.1080/15216540701488358. [PubMed]
- 14. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011; 50:5566–82. https://doi.org/10.1021/bi200642e. [PubMed]
- 15. Cyclin-dependent kinases: inhibition and substrate recognition. Curr Opin Struct Biol. 1999; 9:738–44. https://doi.org/10.1016/s0959-440x(99)00038-x. [PubMed]
- 16. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999; 22:44–52. https://doi.org/10.1038/8751. [PubMed]
- 17. Germ line transmission of the Cdk4(R24C) mutation facilitates tumorigenesis and escape from cellular senescence. Mol Cell Biol. 2002; 22:644–56. https://doi.org/10.1128/MCB.22.2.644-656.2002. [PubMed]
- 18. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997; 11:847–62. https://doi.org/10.1101/gad.11.7.847. [PubMed]
- 19. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999; 18:1571–83. https://doi.org/10.1093/emboj/18.6.1571. [PubMed]
- 20. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008; 14:159–69. https://doi.org/10.1016/j.devcel.2008.01.013. [PubMed]
- 21. Cip/Kip cyclin-dependent protein kinase inhibitors and the road to polyploidy. Cell Div. 2009; 4:10. https://doi.org/10.1186/1747-1028-4-10. [PubMed]
- 22. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol Chem. 1997; 272:25863–72. https://doi.org/10.1074/jbc.272.41.25863. [PubMed]
- 23. Formation of p27-CDK complexes during the human mitotic cell cycle. Cell Growth Differ. 1996; 7:135–46. [PubMed]
- 24. Active cdk6 complexes are predominantly nuclear and represent only a minority of the cdk6 in T cells. Oncogene. 1998; 16:603–11. https://doi.org/10.1038/sj.onc.1201570. [PubMed]
- 25. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol. 2008; 28:498–510. https://doi.org/10.1128/MCB.02171-06. [PubMed]
- 26. Activation of cyclin D1-kinase in murine fibroblasts lacking both p21(Cip1) and p27(Kip1). Oncogene. 2002; 21:8067–74. https://doi.org/10.1038/sj.onc.1206019. [PubMed]
- 27. P27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Mol Cell Biol. 2003; 23:7285–90. https://doi.org/10.1128/MCB.23.20.7285-7290.2003. [PubMed]
- 28. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994; 78:67–74. https://doi.org/10.1016/0092-8674(94)90573-8. [PubMed]
- 29. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994; 78:59–66. https://doi.org/10.1016/0092-8674(94)90572-x. [PubMed]
- 30. Phosphorylation of p27Kip1 regulates assembly and activation of cyclin D1-Cdk4. Mol Cell Biol. 2008; 28:6462–72. https://doi.org/10.1128/MCB.02300-07. [PubMed]
- 31. Analysis of cyclin D3-cdk4 complexes in fibroblasts expressing and lacking p27(kip1) and p21(cip1). Mol Cell Biol. 2000; 20:8748–57. https://doi.org/10.1128/MCB.20.23.8748-8757.2000. [PubMed]
- 32. Altered p27(Kip1) phosphorylation, localization, and function in human epithelial cells resistant to transforming growth factor beta-mediated G(1) arrest. Mol Cell Biol. 2002; 22:2993–3002. https://doi.org/10.1128/MCB.22.9.2993-3002.2002. [PubMed]
- 33. Inactivation of the cyclin D-dependent kinase in the rat fibroblast cell line, 3Y1, induced by contact inhibition. J Biol Chem. 1997; 272:8065–70. https://doi.org/10.1074/jbc.272.12.8065. [PubMed]
- 34. Regulated activating Thr172 phosphorylation of cyclin-dependent kinase 4(CDK4): its relationship with cyclins and CDK “inhibitors”. Mol Cell Biol. 2006; 26:5070–85. https://doi.org/10.1128/MCB.02006-05. [PubMed]
- 35. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996; 15:7060–69. [PubMed]
- 36. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell. 1997; 8:287–301. https://doi.org/10.1091/mbc.8.2.287. [PubMed]
- 37. Defining the substrate specificity of cdk4 kinase-cyclin D1 complex. Carcinogenesis. 1999; 20:193–98. https://doi.org/10.1093/carcin/20.2.193. [PubMed]
- 38. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998; 391:597–601. https://doi.org/10.1038/35404. [PubMed]
- 39. Rb interacts with histone deacetylase to repress transcription. Cell. 1998; 92:463–73. https://doi.org/10.1016/s0092-8674(00)80940-x. [PubMed]
- 40. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998; 391:601–5. https://doi.org/10.1038/35410. [PubMed]
- 41. Targets of cyclin-dependent protein kinases. Curr Opin Cell Biol. 1993; 5:187–93. https://doi.org/10.1016/0955-0674(93)90101-u. [PubMed]
- 42. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004; 430:226–31. https://doi.org/10.1038/nature02650. [PubMed]
- 43. Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J Biol Chem. 2004; 279:17283–88. https://doi.org/10.1074/jbc.C300549200. [PubMed]
- 44. Phosphorylation of the myristoylated protein kinase C substrate MARCKS by the cyclin E-cyclin-dependent kinase 2 complex in vitro. Biochem J. 1999; 340:775–82. [PubMed]
- 45. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011; 20:620–34. https://doi.org/10.1016/j.ccr.2011.10.001. [PubMed]
- 46. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010; 18:329–40. https://doi.org/10.1016/j.ccr.2010.08.012. [PubMed]
- 47. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992; 71:323–34. https://doi.org/10.1016/0092-8674(92)90360-o. [PubMed]
- 48. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993; 73:487–97. https://doi.org/10.1016/0092-8674(93)90136-e. [PubMed]
- 49. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993; 7:331–42. https://doi.org/10.1101/gad.7.3.331. [PubMed]
- 50. CDK4: A Key Player in the Cell Cycle, Development, and Cancer. Genes Cancer. 2012; 3:658–69. https://doi.org/10.1177/1947601913478972. [PubMed]
- 51. The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc Natl Acad Sci U S A. 2009; 106:4171–76. https://doi.org/10.1073/pnas.0809674106. [PubMed]
- 52. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci U S A. 2012; 109:3826–31. https://doi.org/10.1073/pnas.1115201109. [PubMed]
- 53. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007; 448:811–15. https://doi.org/10.1038/nature06046. [PubMed]
- 54. Cdk1, but not Cdk2, is the sole Cdk that is essential and sufficient to drive resumption of meiosis in mouse oocytes. Hum Mol Genet. 2012; 21:2476–84. https://doi.org/10.1093/hmg/dds061. [PubMed]
- 55. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003; 35:25–31. https://doi.org/10.1038/ng1232. [PubMed]
- 56. Cdk2 knockout mice are viable. Curr Biol. 2003; 13:1775–85. https://doi.org/10.1016/j.cub.2003.09.024. [PubMed]
- 57. A premature-termination mutation in the Mus musculus cyclin-dependent kinase 3 gene. Proc Natl Acad Sci U S A. 2001; 98:1682–86. https://doi.org/10.1073/pnas.98.4.1682. [PubMed]
- 58. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999; 19:7011–19. https://doi.org/10.1128/MCB.19.10.7011. [PubMed]
- 59. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004; 118:493–504. https://doi.org/10.1016/j.cell.2004.08.002. [PubMed]
- 60. Mice thrive without Cdk4 and Cdk2. Mol Oncol. 2007; 1:72–83. https://doi.org/10.1016/j.molonc.2007.03.001. [PubMed]
- 61. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004; 118:477–91. https://doi.org/10.1016/j.cell.2004.07.025. [PubMed]
- 62. CDK6 kinase activity is required for thymocyte development. Blood. 2011; 117:6120–31. https://doi.org/10.1182/blood-2010-08-300517. [PubMed]
- 63. A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009; 69:810–18. https://doi.org/10.1158/0008-5472.CAN-08-2473. [PubMed]
- 64. Pituitary hypoplasia and lactotroph dysfunction in mice deficient for cyclin-dependent kinase-4. Endocrinology. 2002; 143:3001–8. https://doi.org/10.1210/endo.143.8.8956. [PubMed]
- 65. Intact follicular maturation and defective luteal function in mice deficient for cyclin-dependent kinase-4. Endocrinology. 2002; 143:647–54. https://doi.org/10.1210/endo.143.2.8611. [PubMed]
- 66. Cdk4 is indispensable for postnatal proliferation of the anterior pituitary. J Biol Chem. 2004; 279:51100–6. https://doi.org/10.1074/jbc.M409080200. [PubMed]
- 67. Characterization of the abnormal pancreatic development, reduced growth and infertility in Cdk4 mutant mice. Oncogene. 2003; 22:8413–21. https://doi.org/10.1038/sj.onc.1206888. [PubMed]
- 68. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene. 2003; 22:5261–69. https://doi.org/10.1038/sj.onc.1206506. [PubMed]
- 69. Cdk4 promotes adipogenesis through PPARgamma activation. Cell Metab. 2005; 2:239–49. https://doi.org/10.1016/j.cmet.2005.09.003. [PubMed]
- 70. Role of Cdk4 in lymphocyte function and allergen response. Cell Cycle. 2010; 9:4922–30. https://doi.org/10.4161/cc.9.24.14209. [PubMed]
- 71. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993; 8:2127–33. [PubMed]
- 72. Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Lett. 1995; 90:43–50. https://doi.org/10.1016/0304-3835(94)03676-a. [PubMed]
- 73. D11S287, a putative oncogene on chromosome 11q13, is amplified and expressed in squamous cell and mammary carcinomas and linked to BCL-1. Oncogene. 1991; 6:439–44. [PubMed]
- 74. Cyclin D1 and prognosis in human breast cancer. Int J Cancer. 1996; 69:92–99. https://doi.org/10.1002/(SICI)1097-0215(19960422)69:2<92::AID-IJC4>3.0.CO;2-Q. [PubMed]
- 75. Determination of the prognostic value of cyclin D1 overexpression in breast cancer. Oncogene. 1995; 11:885–91. [PubMed]
- 76. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer. 1994; 57:353–61. https://doi.org/10.1002/ijc.2910570311. [PubMed]
- 77. Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinomas from non-malignant lesions. Nat Med. 1995; 1:1257–60. https://doi.org/10.1038/nm1295-1257. [PubMed]
- 78. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005; 65:10174–78. https://doi.org/10.1158/0008-5472.CAN-05-2639. [PubMed]
- 79. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001; 411:1017–21. https://doi.org/10.1038/35082500. [PubMed]
- 80. The role of the cyclin D1dependent kinases in ErbB2-mediated breast cancer. Am J Pathol. 2004; 164:1031–38. https://doi.org/10.1016/S0002-9440(10)63190-2. [PubMed]
- 81. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006; 9:13–22. https://doi.org/10.1016/j.ccr.2005.12.019. [PubMed]
- 82. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006; 9:23–32. https://doi.org/10.1016/j.ccr.2005.12.012. [PubMed]
- 83. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell. 2003; 114:323–34. https://doi.org/10.1016/s0092-8674(03)00570-1. [PubMed]
- 84. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995; 269:1281–84. https://doi.org/10.1126/science.7652577. [PubMed]
- 85. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996; 12:97–99. https://doi.org/10.1038/ng0196-97. [PubMed]
- 86. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. EMBO J. 2001; 20:6637–47. https://doi.org/10.1093/emboj/20.23.6637. [PubMed]
- 87. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996; 85:27–37. https://doi.org/10.1016/s0092-8674(00)81079-x. [PubMed]
- 88. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995; 82:675–84. https://doi.org/10.1016/0092-8674(95)90039-x. [PubMed]
- 89. The absence of p21Cip1/WAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes Dev. 1996; 10:3065–75. https://doi.org/10.1101/gad.10.23.3065. [PubMed]
- 90. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998; 12:2899–911. https://doi.org/10.1101/gad.12.18.2899. [PubMed]
- 91. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996; 85:733–44. https://doi.org/10.1016/s0092-8674(00)81239-8. [PubMed]
- 92. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell. 1996; 85:721–32. https://doi.org/10.1016/s0092-8674(00)81238-6. [PubMed]
- 93. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996; 85:707–20. https://doi.org/10.1016/s0092-8674(00)81237-4. [PubMed]
- 94. HMGA2 cooperates with either p27kip1 deficiency or Cdk4R24C mutation in pituitary tumorigenesis. Cell Cycle. 2018; 17:580–88. https://doi.org/10.1080/15384101.2017.1403682. [PubMed]
- 95. Invasive melanoma in Cdk4-targeted mice. Proc Natl Acad Sci U S A. 2001; 98:13312–17. https://doi.org/10.1073/pnas.241338598. [PubMed]
- 96. Cooperativity of Cdk4R24C and Ras in melanoma development. Cell Cycle. 2010; 9:3305–14. https://doi.org/10.4161/cc.9.16.12632. [PubMed]
- 97. Spontaneous and UV radiation-induced multiple metastatic melanomas in Cdk4R24C/R24C/TPras mice. Cancer Res. 2006; 66:2946–52. https://doi.org/10.1158/0008-5472.CAN-05-3196. [PubMed]
- 98. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999; 400:468–72. https://doi.org/10.1038/22788. [PubMed]
- 99. Quantitation of early clonal expansion of two mutant 61st codon c-Ha-ras alleles in DMBA/TPA treated mouse skin by nested PCR/RFLP. Carcinogenesis. 1996; 17:2551–57. https://doi.org/10.1093/carcin/17.12.2551. [PubMed]
- 100. Cdk4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am J Pathol. 2002; 161:405–11. https://doi.org/10.1016/S0002-9440(10)64196-X. [PubMed]
- 101. Rapid growth of invasive metastatic melanoma in carcinogen-treated hepatocyte growth factor/scatter factor-transgenic mice carrying an oncogenic CDK4 mutation. Am J Pathol. 2006; 169:665–72. https://doi.org/10.2353/ajpath.2006.060017. [PubMed]
- 102. Neonatal UVB exposure accelerates melanoma growth and enhances distant metastases in Hgf-Cdk4(R24C) C57BL/6 mice. Int J Cancer. 2011; 129:285–94. https://doi.org/10.1002/ijc.25913. [PubMed]
- 103. Loss of nuclear receptor RXRα in epidermal keratinocytes promotes the formation of Cdk4-activated invasive melanomas. Pigment Cell Melanoma Res. 2010; 23:635–48. https://doi.org/10.1111/j.1755-148X.2010.00732.x. [PubMed]
- 104. Increased angiogenesis in Cdk4(R24C/R24C):Apc(+/Min) intestinal tumors. Cell Cycle. 2010; 9:2456–63. https://doi.org/10.4161/cc.9.12.12055. [PubMed]
- 105. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994; 369:669–71. https://doi.org/10.1038/369669a0. [PubMed]
- 106. Requirement of Cdk4 for v-Ha-ras-Induced Breast Tumorigenesis and Activation of the v-ras-Induced Senescence Program by the R24C Mutation. Genes Cancer. 2010; 1:69–80. https://doi.org/10.1177/1947601909358105. [PubMed]
- 107. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010; 18:63–73. https://doi.org/10.1016/j.ccr.2010.05.025. [PubMed]
- 108. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014; 13:828–51. https://doi.org/10.1038/nrd4389. [PubMed]
- 109. CDK4 regulates cancer stemness and is a novel therapeutic target for triple-negative breast cancer. Sci Rep. 2016; 6:35383. https://doi.org/10.1038/srep35383. [PubMed]
- 110. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci U S A. 2007; 104:10703–8. https://doi.org/10.1073/pnas.0611568104. [PubMed]
- 111. Cyclin D3 promotes adipogenesis through activation of peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2005; 25:9985–95. https://doi.org/10.1128/MCB.25.22.9985-9995.2005. [PubMed]
- 112. Peroxisome proliferator-activated receptor gamma recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol Endocrinol. 2006; 20:1494–505. https://doi.org/10.1210/me.2005-0222. [PubMed]
- 113. Metabolic control in cancer cells. Ann Endocrinol (Paris). 2013; 74:71–73. https://doi.org/10.1016/j.ando.2013.03.021. [PubMed]
- 114. CDK4 is an essential insulin effector in adipocytes. J Clin Invest. 2016; 126:335–48. https://doi.org/10.1172/JCI81480. [PubMed]
- 115. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014; 510:547–51. https://doi.org/10.1038/nature13267. [PubMed]
- 116. CDK4 Phosphorylates AMPKα2 to Inhibit Its Activity and Repress Fatty Acid Oxidation. Mol Cell. 2017; 68:336–49.e6. https://doi.org/10.1016/j.molcel.2017.09.034. [PubMed]
- 117. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature. 2017; 546:426–30. https://doi.org/10.1038/nature22797. [PubMed]
- 118. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005; 24:2909–15. https://doi.org/10.1038/sj.onc.1208618. [PubMed]
- 119. Cyclin-dependent kinase 4/6 (cdk4/6) inhibitors: perspectives in cancer therapy and imaging. Mini Rev Med Chem. 2010; 10:527–39. https://doi.org/10.2174/138955710791384072. [PubMed]
- 120. Disruption of the cyclin D/cyclin-dependent kinase/INK4/retinoblastoma protein regulatory pathway in human neuroblastoma. Cancer Res. 1998; 58:2624–32. [PubMed]
- 121. Dysregulation of cyclin dependent kinase 6 expression in splenic marginal zone lymphoma through chromosome 7q translocations. Oncogene. 1999; 18:6271–77. https://doi.org/10.1038/sj.onc.1203033. [PubMed]
- 122. In B-cell chronic lymphocytic leukemias, 7q21 translocations lead to overexpression of the CDK6 gene. Blood. 2003; 102:1549–50. https://doi.org/10.1182/blood-2003-04-1220. [PubMed]
- 123. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002; 1602:73–87. https://doi.org/10.1016/s0304-419x(02)00037-9. [PubMed]
- 124. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008; 100:672–79. https://doi.org/10.1093/jnci/djn123. [PubMed]
- 125. Cancer stem cells: an old idea--a paradigm shift. Cancer Res. 2006; 66:1883. https://doi.org/10.1158/0008-5472.CAN-05-3153. [PubMed]
- 126. Targeting tumour-initiating cells to improve the cure rates for triple-negative breast cancer. Expert Rev Mol Med. 2010; 12:e22. https://doi.org/10.1017/S1462399410001535. [PubMed]
- 127. Breast cancer stem cells: treatment resistance and therapeutic opportunities. Carcinogenesis. 2011; 32:650–58. https://doi.org/10.1093/carcin/bgr028. [PubMed]
- 128. BMP4 administration induces differentiation of CD133+ hepatic cancer stem cells, blocking their contributions to hepatocellular carcinoma. Cancer Res. 2012; 72:4276–85. https://doi.org/10.1158/0008-5472.CAN-12-1013. [PubMed]
- 129. A possible usage of a CDK4 inhibitor for breast cancer stem cell-targeted therapy. Biochem Biophys Res Commun. 2013; 430:1329–33. https://doi.org/10.1016/j.bbrc.2012.10.119. [PubMed]
- 130. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011; 11:558–72. https://doi.org/10.1038/nrc3090. [PubMed]
- 131. Targeting cyclins and cyclin-dependent kinases in cancer: lessons from mice, hopes for therapeutic applications in human. Cell Cycle. 2006; 5:2110–14. https://doi.org/10.4161/cc.5.18.3218. [PubMed]
- 132. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004; 3:1427–38. [PubMed]
- 133. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005; 48:2388–406. https://doi.org/10.1021/jm049354h. [PubMed]
- 134. Pharmacologic inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Mol Cancer Ther. 2006; 5:1299–308. https://doi.org/10.1158/1535-7163.MCT-05-0383. [PubMed]
- 135. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006; 66:7661–67. https://doi.org/10.1158/0008-5472.CAN-06-1098. [PubMed]
- 136. Cell cycle and biochemical effects of PD 0183812. A potent inhibitor of the cyclin D-dependent kinases CDK4 and CDK6. J Biol Chem. 2001; 276:16617–23. https://doi.org/10.1074/jbc.M008867200. [PubMed]
- 137. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood. 2006; 108:1744–50. https://doi.org/10.1182/blood-2006-04-016634. [PubMed]
- 138. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009; 11:R77. https://doi.org/10.1186/bcr2419. [PubMed]
- 139. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010; 70:3228–38. https://doi.org/10.1158/0008-5472.CAN-09-4559. [PubMed]
- 140. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci U S A. 2010; 107:11501–6. https://doi.org/10.1073/pnas.1001613107. [PubMed]
- 141. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res. 2011; 17:1591–602. https://doi.org/10.1158/1078-0432.CCR-10-2307. [PubMed]
- 142. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011; 104:1862–68. https://doi.org/10.1038/bjc.2011.177. [PubMed]
- 143. A phase I dose escalation trial of a daily oral CDK 4/6 inhibitor PD-0332991. J Clin Oncol. 2007; 25:3550. https://doi.org/10.1200/jco.2007.25.18_suppl.3550.
- 144. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012; 18:568–76. https://doi.org/10.1158/1078-0432.CCR-11-0509. [PubMed]
- 145. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016; 17:425–39. https://doi.org/10.1016/S1470-2045(15)00613-0. [PubMed]
- 146. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N Engl J Med. 2018; 379:1926–36. https://doi.org/10.1056/NEJMoa1810527. [PubMed]
- 147. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015; 14:130–46. https://doi.org/10.1038/nrd4504. [PubMed]
- 148. The Role of CDK4/6 Inhibition in Breast Cancer. Oncologist. 2015; 20:483–90. https://doi.org/10.1634/theoncologist.2014-0443. [PubMed]
- 149. Arrested Developments: CDK4/6 Inhibitor Resistance and Alterations in the Tumor Immune Microenvironment. Clin Cancer Res. 2019; 25:921–27. https://doi.org/10.1158/1078-0432.CCR-18-1967. [PubMed]
- 150. Abstract PR02: LEE011: an orally bioavailable, selective small molecule inhibitor of CDK4/6– Reactivating Rb in cancer. Mol Cancer Ther. 2013; 12:PR02. https://doi.org/10.1158/1535-7163.TARG-13-PR02.
- 151. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med. 2016; 375:1738–48. https://doi.org/10.1056/NEJMoa1609709. [PubMed]
- 152. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016; 6:353–67. https://doi.org/10.1158/2159-8290.CD-15-0894. [PubMed]
- 153. Inhibiting CDK in Cancer Therapy: Current Evidence and Future Directions. Target Oncol. 2018; 13:21–38. https://doi.org/10.1007/s11523-017-0541-2. [PubMed]
- 154. Ribociclib for post-menopausal women with HR+/HER2-advanced or metastatic breast cancer. Expert Rev Clin Pharmacol. 2017; 10:1169–76. https://doi.org/10.1080/17512433.2017.1376653. [PubMed]
- 155. Abemaciclib Combined With Endocrine Therapy for the Adjuvant Treatment of HR+, HER2-, Node-Positive, High-Risk, Early Breast Cancer (monarchE). J Clin Oncol. 2020; 38:3987–98. https://doi.org/10.1200/JCO.20.02514. [PubMed]
- 156. Palbociclib with adjuvant endocrine therapy in early breast cancer (PALLAS): interim analysis of a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2021; 22:212–22. https://doi.org/10.1016/S1470-2045(20)30642-2. [PubMed]
- 157. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008; 416:375–85. https://doi.org/10.1042/BJ20081668. [PubMed]
- 158. Inhibition of CK1ε potentiates the therapeutic efficacy of CDK4/6 inhibitor in breast cancer. Nat Commun. 2021; 12:5386. https://doi.org/10.1038/s41467-021-25700-6. [PubMed]
- 159. Combined CDK4/6 and PI3Kα Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 2017; 77:6340–52. https://doi.org/10.1158/0008-5472.CAN-17-2210. [PubMed]
- 160. Short-term CDK4/6 Inhibition Radiosensitizes Estrogen Receptor-Positive Breast Cancers. Clin Cancer Res. 2020; 26:6568–80. https://doi.org/10.1158/1078-0432.CCR-20-2269. [PubMed]
- 161. The Ongoing Search for Biomarkers of CDK4/6 Inhibitor Responsiveness in Breast Cancer. Mol Cancer Ther. 2020; 19:3–12. https://doi.org/10.1158/1535-7163.MCT-19-0253. [PubMed]
- 162. Accessed July, 2022.
- 163. Mechanistic Investigation of Bone Marrow Suppression Associated with Palbociclib and its Differentiation from Cytotoxic Chemotherapies. Clin Cancer Res. 2016; 22:2000–8. https://doi.org/10.1158/1078-0432.CCR-15-1421. [PubMed]
- 164. .
- 165. .
- 166. .
- 167. Accessed February 2022.
- 168. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci Rep. 2015; 5:13110. https://doi.org/10.1038/srep13110. [PubMed]
- 169. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015; 27:450–61. https://doi.org/10.1016/j.ccell.2015.03.001. [PubMed]
- 170. Immunotherapy for Breast Cancer: What Are We Missing? Clin Cancer Res. 2017; 23:2640–46. https://doi.org/10.1158/1078-0432.CCR-16-2569. [PubMed]
- 171. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017; 548:471–75. https://doi.org/10.1038/nature23465. [PubMed]
- 172. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018; 8:216–33. https://doi.org/10.1158/2159-8290.CD-17-0915. [PubMed]
- 173. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018; 553:91–95. https://doi.org/10.1038/nature25015. [PubMed]
- 174. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018; 22:2978–94. https://doi.org/10.1016/j.celrep.2018.02.053. [PubMed]
- 175. CDK4/6 Inhibition Promotes Antitumor Immunity through the Induction of T-cell Memory. Cancer Discov. 2021; 11:2582–601. https://doi.org/10.1158/2159-8290.CD-20-1554. [PubMed]
- 176. Inhibition of CDK4/6 Promotes CD8 T-cell Memory Formation. Cancer Discov. 2021; 11:2564–81. https://doi.org/10.1158/2159-8290.CD-20-1540. [PubMed]
- 177. Phase I/II trial of palbociclib, pembrolizumab and letrozole in patients with hormone receptor-positive metastatic breast cancer. Eur J Cancer. 2021; 154:11–20. https://doi.org/10.1016/j.ejca.2021.05.035. [PubMed]
- 178. CDK4 deficiency promotes genomic instability and enhances Myc-driven lymphomagenesis. J Clin Invest. 2014; 124:1672–84. https://doi.org/10.1172/JCI63139. [PubMed]
- 179. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2013; 24:167–81. https://doi.org/10.1016/j.ccr.2013.07.012. [PubMed]
- 180. Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front Pharmacol. 2020; 11:580251. https://doi.org/10.3389/fphar.2020.580251. [PubMed]
- 181. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013; 45:1446–51. https://doi.org/10.1038/ng.2823. [PubMed]
- 182. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013; 45:1439–45. https://doi.org/10.1038/ng.2822. [PubMed]
- 183. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013; 73:6856–64. https://doi.org/10.1158/0008-5472.CAN-13-1197. [PubMed]
- 184. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014; 20:1757–67. https://doi.org/10.1158/1078-0432.CCR-13-2332. [PubMed]
- 185. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J Clin Oncol. 2016; 34:2961–68. https://doi.org/10.1200/JCO.2016.67.3061. [PubMed]
- 186. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat Genet. 2019; 51:207–16. https://doi.org/10.1038/s41588-018-0287-5. [PubMed]
- 187. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016; 76:2301–13. https://doi.org/10.1158/0008-5472.CAN-15-0728. [PubMed]
- 188. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019; 10:1373. https://doi.org/10.1038/s41467-019-09068-2. [PubMed]
- 189. Activation of the IFN Signaling Pathway is Associated with Resistance to CDK4/6 Inhibitors and Immune Checkpoint Activation in ER-Positive Breast Cancer. Clin Cancer Res. 2021; 27:4870–82. https://doi.org/10.1158/1078-0432.CCR-19-4191. [PubMed]
- 190. Mechanisms of Resistance to CDK4/6 Inhibitors in Breast Cancer and Potential Biomarkers of Response. Breast Care (Basel). 2017; 12:304–8. https://doi.org/10.1159/000484167. [PubMed]
- 191. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr Relat Cancer. 2019; 26:R15–30. https://doi.org/10.1530/ERC-18-0317. [PubMed]
- 192. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell. 2020; 37:514–29. https://doi.org/10.1016/j.ccell.2020.03.010. [PubMed]
- 193. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017; 77:2488–99. https://doi.org/10.1158/0008-5472.CAN-16-2653. [PubMed]
- 194. Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep. 2020; 31:107504. https://doi.org/10.1016/j.celrep.2020.03.068. [PubMed]
- 195. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-positive Breast Cancer. Mol Cancer Ther. 2018; 17:908–20. https://doi.org/10.1158/1535-7163.MCT-17-0537. [PubMed]
- 196. Everolimus Reverses Palbociclib Resistance in ER+ Human Breast Cancer Cells by Inhibiting Phosphatidylinositol 3-Kinase(PI3K)/Akt/Mammalian Target of Rapamycin (mTOR) Pathway. Med Sci Monit. 2019; 25:77–86. https://doi.org/10.12659/MSM.912929. [PubMed]
- 197. Sequencing Endocrine Therapy for Metastatic Breast Cancer: What Do We Do After Disease Progression on a CDK4/6 Inhibitor? Curr Oncol Rep. 2020; 22:57. https://doi.org/10.1007/s11912-020-00917-8. [PubMed]
- 198. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science. 2019; 366:eaaw2106. https://doi.org/10.1126/science.aaw2106. [PubMed]
- 199. Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin D-CDK4 to inhibit CDK2. Nat Commun. 2021; 12:3356. https://doi.org/10.1038/s41467-021-23612-z. [PubMed]
- 200. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell. 2017; 32:761–76.e6. https://doi.org/10.1016/j.ccell.2017.11.006. [PubMed]
- 201. The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget. 2018; 9:35226–40. https://doi.org/10.18632/oncotarget.26215. [PubMed]
- 202. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat Cancer. 2021; 2:429–43. https://doi.org/10.1038/s43018-021-00174-z. [PubMed]
- 203. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell. 2020; 181:102–14. https://doi.org/10.1016/j.cell.2019.11.031. [PubMed]
- 204. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem Commun (Camb). 2019; 55:2704–7. https://doi.org/10.1039/c9cc00163h. [PubMed]
- 205. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew Chem Int Ed Engl. 2019; 58:6321–26. https://doi.org/10.1002/anie.201901336. [PubMed]
- 206. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J Med Chem. 2019; 62:7575–82. https://doi.org/10.1021/acs.jmedchem.9b00871. [PubMed]
- 207. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem Biol. 2019; 26:300–6.e9. https://doi.org/10.1016/j.chembiol.2018.11.006. [PubMed]
- 208. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem Sci. 2020; 11:3474–86. https://doi.org/10.1039/d0sc00167h. [PubMed]
- 209. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg Med Chem Lett. 2020; 30:127106. https://doi.org/10.1016/j.bmcl.2020.127106. [PubMed]
- 210. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood. 2020; 135:1560–73. https://doi.org/10.1182/blood.2019003604. [PubMed]
- 211. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell. 2021; 39:1404–21.e11. https://doi.org/10.1016/j.ccell.2021.08.009. [PubMed]
- 212. CCND1-CDK4-mediated cell cycle progression provides a competitive advantage for human hematopoietic stem cells in vivo. J Exp Med. 2015; 212:1171–83. https://doi.org/10.1084/jem.20150308. [PubMed]
- 213. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med. 2017; 9:eaal3986. https://doi.org/10.1126/scitranslmed.aal3986. [PubMed]
- 214. Preclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-Induced Myelosuppression. Mol Cancer Ther. 2016; 15:783–93. https://doi.org/10.1158/1535-7163.MCT-15-0775. [PubMed]
 All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.